

Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells
- PMID: 24280423
- PMCID: PMC4178217
- DOI: 10.1186/2049-3002-1-19
Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells
Abstract
Background: Most normal cells in the presence of oxygen utilize glucose for mitochondrial oxidative phosphorylation. In contrast, many cancer cells rapidly convert glucose to lactate in the cytosol, a process termed aerobic glycolysis. This glycolytic phenotype is enabled by lactate dehydrogenase (LDH), which catalyzes the inter-conversion of pyruvate and lactate. The purpose of this study was to identify and characterize potent and selective inhibitors of LDHA.
Methods: High throughput screening and lead optimization were used to generate inhibitors of LDHA enzymatic activity. Effects of these inhibitors on metabolism were evaluated using cell-based lactate production, oxygen consumption, and 13C NMR spectroscopy assays. Changes in comprehensive metabolic profile, cell proliferation, and apoptosis were assessed upon compound treatment.
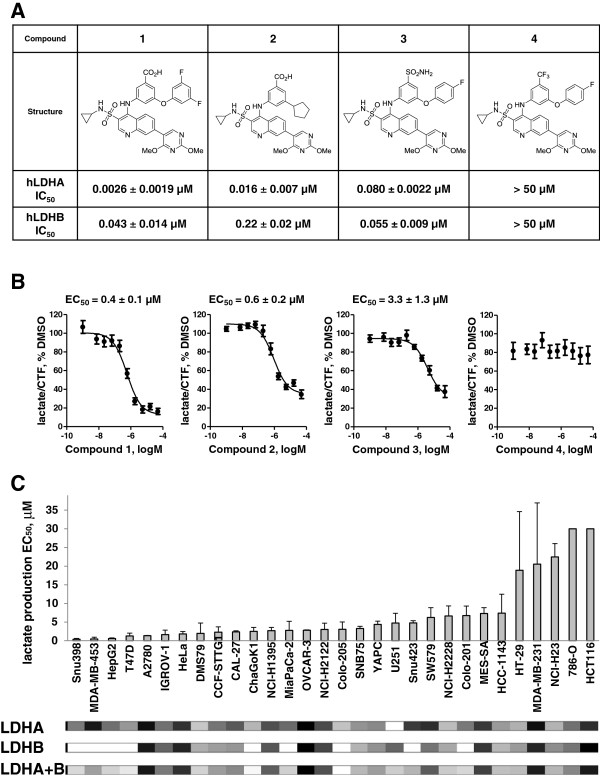
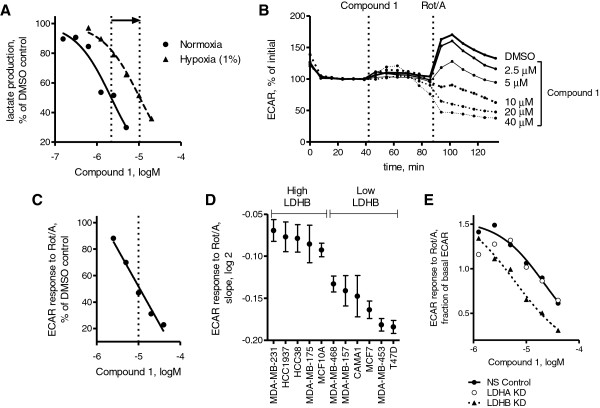
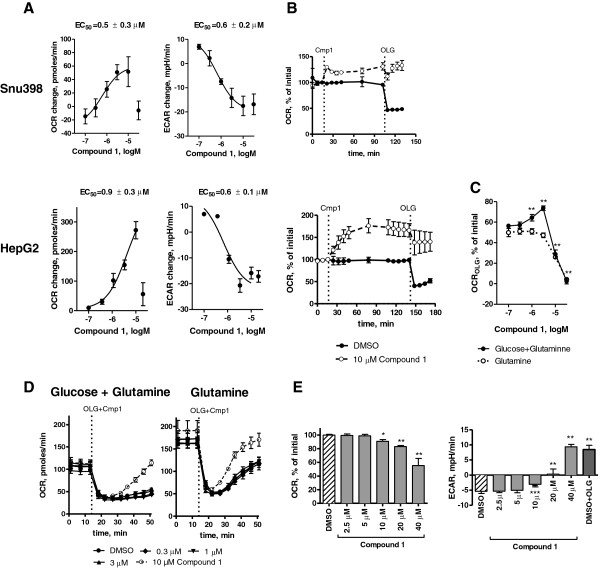
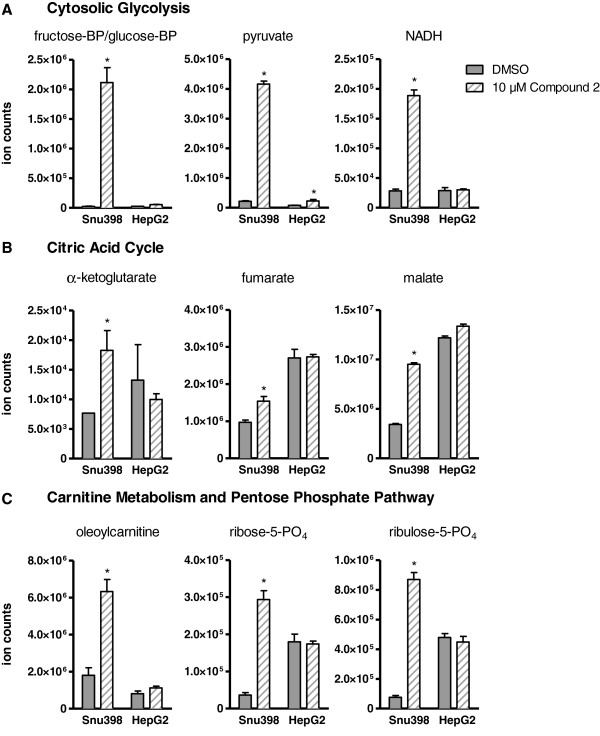
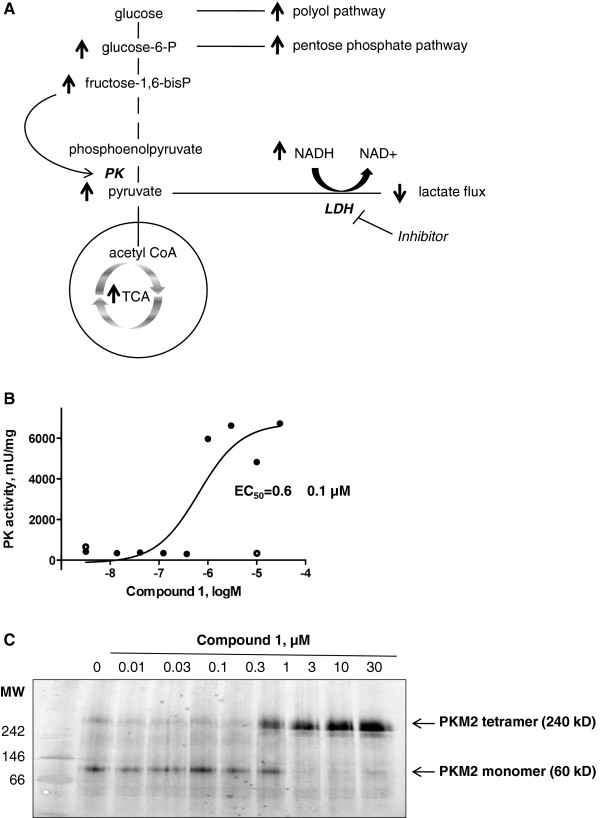
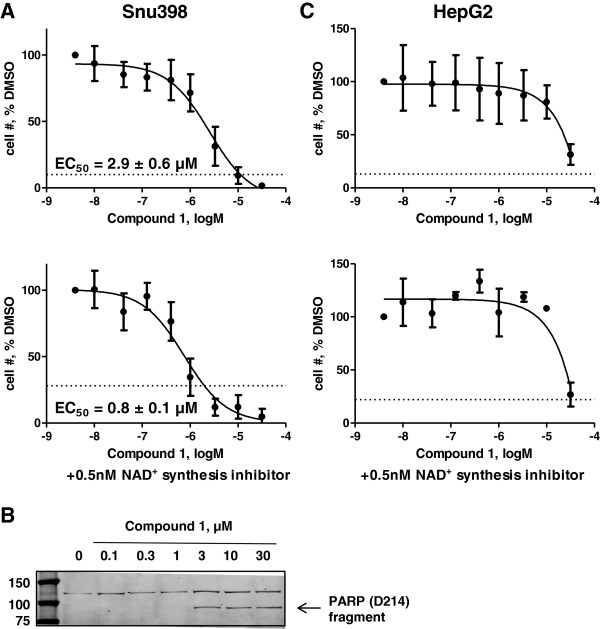
Results: 3-((3-carbamoyl-7-(3,5-dimethylisoxazol-4-yl)-6-methoxyquinolin-4-yl) amino) benzoic acid was identified as an NADH-competitive LDHA inhibitor. Lead optimization yielded molecules with LDHA inhibitory potencies as low as 2 nM and 10 to 80-fold selectivity over LDHB. Molecules in this family rapidly and profoundly inhibited lactate production rates in multiple cancer cell lines including hepatocellular and breast carcinomas. Consistent with selective inhibition of LDHA, the most sensitive breast cancer cell lines to lactate inhibition in hypoxic conditions were cells with low expression of LDHB. Our inhibitors increased rates of oxygen consumption in hepatocellular carcinoma cells at doses up to 3 microM, while higher concentrations directly inhibited mitochondrial function. Analysis of more than 500 metabolites upon LDHA inhibition in Snu398 cells revealed that intracellular concentrations of glycolysis and citric acid cycle intermediates were increased, consistent with enhanced Krebs cycle activity and blockage of cytosolic glycolysis. Treatment with these compounds also potentiated PKM2 activity and promoted apoptosis in Snu398 cells.
Conclusions: Rapid chemical inhibition of LDHA by these quinoline 3-sulfonamids led to profound metabolic alterations and impaired cell survival in carcinoma cells making it a compelling strategy for treating solid tumors that rely on aerobic glycolysis for survival.
Figures

References
-
- Kolev Y, Uetake H, Takagi Y, Sugihara K. Lactate dehydrogenase-5 (LDH-5) expression in human gastric cancer: association with hypoxia-inducible factor (HIF-1alpha) pathway, angiogenic factors production and poor prognosis. Ann Surg Oncol. 2008;15:2336–2344. doi: 10.1245/s10434-008-9955-5. - DOI - PubMed
-
- Koukourakis MI, Giatromanolaki A, Sivridis E, Bougioukas G, Didilis V, Gatter KC, Harris AL. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br J Cancer. 2003;89:877–885. doi: 10.1038/sj.bjc.6601205. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
