

COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylcholinesterase impair the interaction of ColQ with proteins of the basal lamina
- PMID: 24281389
- PMCID: PMC4024244
- DOI: 10.1007/s00439-013-1391-3
COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylcholinesterase impair the interaction of ColQ with proteins of the basal lamina
Abstract
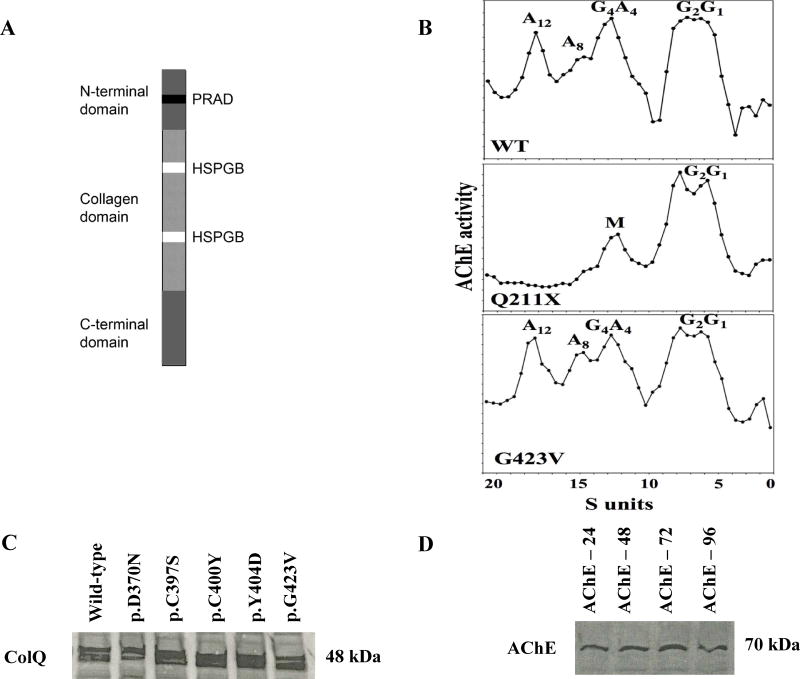
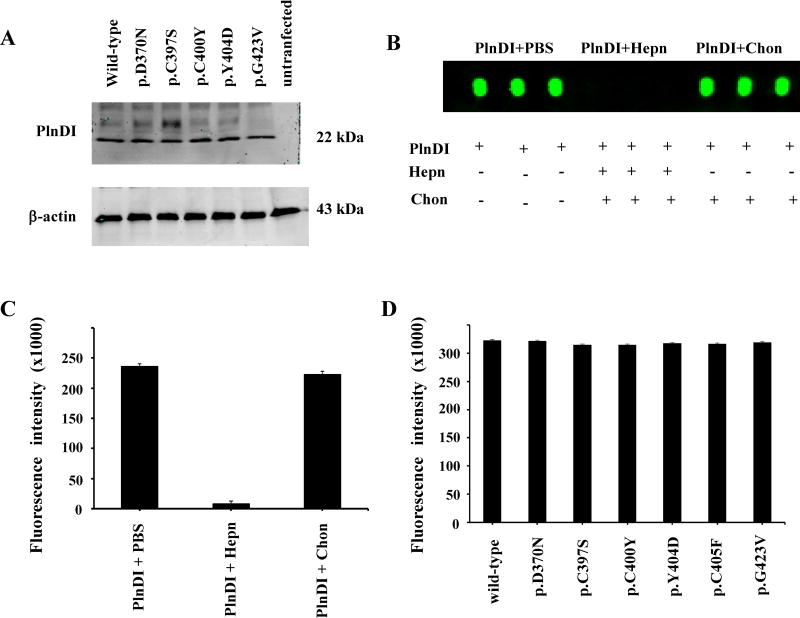
Collagen Q (ColQ) is a key multidomain functional protein of the neuromuscular junction (NMJ), crucial for anchoring acetylcholinesterase (AChE) to the basal lamina (BL) and accumulating AChE at the NMJ. The attachment of AChE to the BL is primarily accomplished by the binding of the ColQ collagen domain to the heparan sulfate proteoglycan perlecan and the COOH-terminus to the muscle-specific receptor tyrosine kinase (MuSK), which in turn plays a fundamental role in the development and maintenance of the NMJ. Yet, the precise mechanism by which ColQ anchors AChE at the NMJ remains unknown. We identified five novel mutations at the COOH-terminus of ColQ in seven patients from five families affected with endplate (EP) AChE deficiency. We found that the mutations do not affect the assembly of ColQ with AChE to form asymmetric forms of AChE or impair the interaction of ColQ with perlecan. By contrast, all mutations impair in varied degree the interaction of ColQ with MuSK as well as basement membrane extract (BME) that have no detectable MuSK. Our data confirm that the interaction of ColQ to perlecan and MuSK is crucial for anchoring AChE to the NMJ. In addition, the identified COOH-terminal mutants not only reduce the interaction of ColQ with MuSK, but also diminish the interaction of ColQ with BME. These findings suggest that the impaired attachment of COOH-terminal mutants causing EP AChE deficiency is in part independent of MuSK, and that the COOH-terminus of ColQ may interact with other proteins at the BL.
Figures







Similar articles
-
Specific binding of collagen Q to the neuromuscular junction is exploited to cure congenital myasthenia and to explore bases of myasthenia gravis.Chem Biol Interact. 2013 Mar 25;203(1):335-40. doi: 10.1016/j.cbi.2012.08.020. Epub 2012 Sep 8. Chem Biol Interact. 2013. PMID: 22981737 Free PMC article.
-
Mutations in the C-terminal domain of ColQ in endplate acetylcholinesterase deficiency compromise ColQ-MuSK interaction.Hum Mutat. 2013 Jul;34(7):997-1004. doi: 10.1002/humu.22325. Epub 2013 Apr 19. Hum Mutat. 2013. PMID: 23553736
-
Developmental consequences of the ColQ/MuSK interactions.Chem Biol Interact. 2013 Mar 25;203(1):287-91. doi: 10.1016/j.cbi.2012.10.006. Epub 2012 Oct 23. Chem Biol Interact. 2013. PMID: 23089045 Review.
-
MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction.J Cell Biol. 2004 May 24;165(4):505-15. doi: 10.1083/jcb.200307164. J Cell Biol. 2004. PMID: 15159418 Free PMC article.
-
Collagen Q is a key player for developing rational therapy for congenital myasthenia and for dissecting the mechanisms of anti-MuSK myasthenia gravis.J Mol Neurosci. 2014 Jul;53(3):359-61. doi: 10.1007/s12031-013-0170-x. J Mol Neurosci. 2014. PMID: 24234034 Review.
Cited by
-
Pharmacological Treatments for Congenital Myasthenic Syndromes Caused by COLQ Mutations.Curr Neuropharmacol. 2023;21(7):1594-1605. doi: 10.2174/1570159X21666230126145652. Curr Neuropharmacol. 2023. PMID: 36703579 Free PMC article. Review.
-
Genome-wide association analyses identify 39 new susceptibility loci for diverticular disease.Nat Genet. 2018 Oct;50(10):1359-1365. doi: 10.1038/s41588-018-0203-z. Epub 2018 Sep 3. Nat Genet. 2018. PMID: 30177863 Free PMC article.
-
Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review.Int J Mol Sci. 2023 Feb 13;24(4):3730. doi: 10.3390/ijms24043730. Int J Mol Sci. 2023. PMID: 36835142 Free PMC article. Review.
-
Molecular Analysis of a Congenital Myasthenic Syndrome Due to a Pathogenic Variant Affecting the C-Terminus of ColQ.Int J Mol Sci. 2023 Nov 11;24(22):16217. doi: 10.3390/ijms242216217. Int J Mol Sci. 2023. PMID: 38003406 Free PMC article.
-
A presynaptic congenital myasthenic syndrome attributed to a homozygous sequence variant in LAMA5.Ann N Y Acad Sci. 2018 Feb;1413(1):119-125. doi: 10.1111/nyas.13585. Epub 2018 Jan 28. Ann N Y Acad Sci. 2018. PMID: 29377152 Free PMC article. Review.
References
-
- Amershan Biosciences. Affinity Chromatography Handbook, Principles and Methods, Code No. 18-1022-29. 2002.
-
- Antolik C, Catino DH, Resneck WG, Bloch RJ. The tetratricopeptide repeat domains of rapsyn bind directly to cytoplasmic sequences of the muscle-specific kinase. Neuroscience. 2006;141:87–100. - PubMed
-
- Arikawa-Hirasawa E, Rossi SG, Rotundo RL, Yamada Y. Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat Neurosci. 2002;5:119–123. - PubMed
-
- Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: acceleration of sequential expression of α5 and α7 nicotinic receptor subunits in oral keratinocytes exposed to cigarette smoke. FASEB J. 2008;22:1356–1368. - PubMed
-
- Arredondo J, Chernyavsky AI, Karaouni A, Jolkosky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006;20:2093–2101. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
