

Fibrinogen species as resolved by HPLC-SAXS data processing within the UltraScan Solution Modeler (US-SOMO) enhanced SAS module
- PMID: 24282333
- PMCID: PMC3831300
- DOI: 10.1107/S0021889813027751
Fibrinogen species as resolved by HPLC-SAXS data processing within the UltraScan Solution Modeler (US-SOMO) enhanced SAS module
Abstract
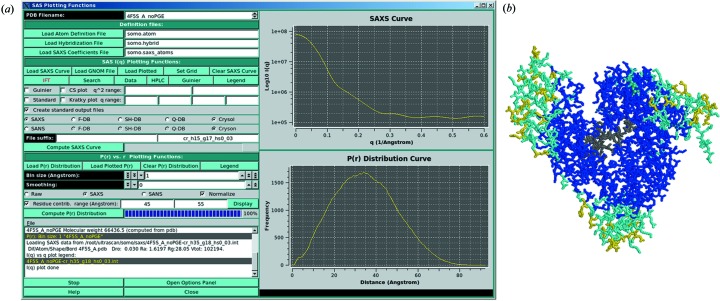
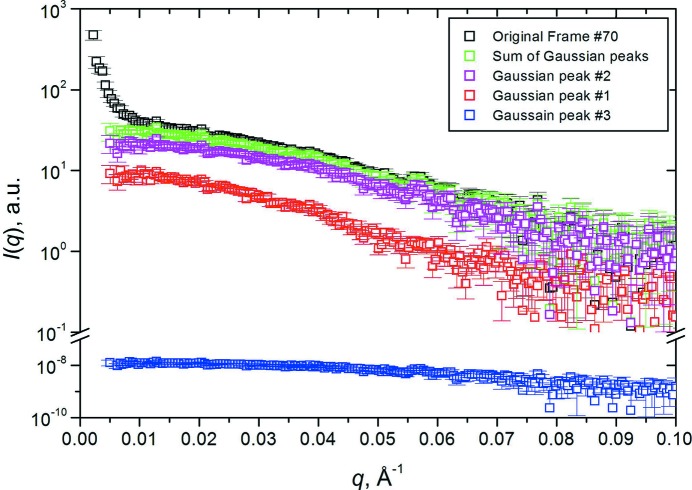
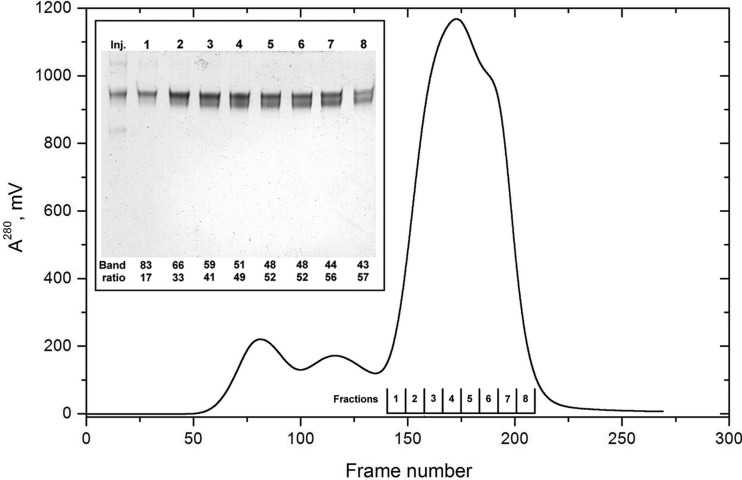
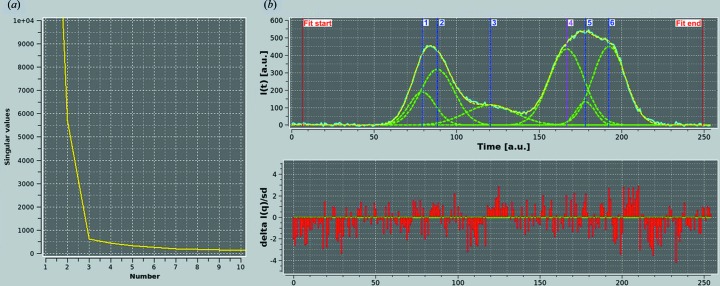
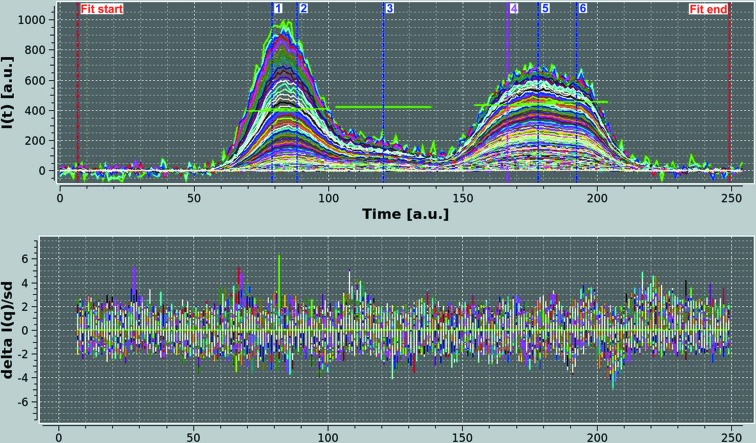
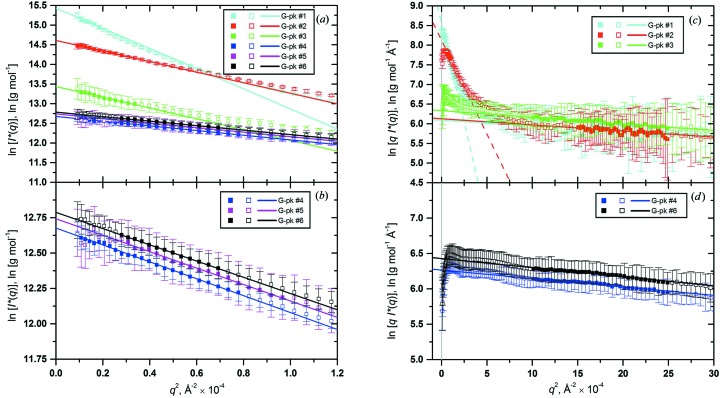
Fibrinogen is a large heterogeneous aggregation/degradation-prone protein playing a central role in blood coagulation and associated pathologies, whose structure is not completely resolved. When a high-molecular-weight fraction was analyzed by size-exclusion high-performance liquid chromatography/small-angle X-ray scattering (HPLC-SAXS), several composite peaks were apparent and because of the stickiness of fibrinogen the analysis was complicated by severe capillary fouling. Novel SAS analysis tools developed as a part of the UltraScan Solution Modeler (US-SOMO; http://somo.uthscsa.edu/), an open-source suite of utilities with advanced graphical user interfaces whose initial goal was the hydrodynamic modeling of biomacromolecules, were implemented and applied to this problem. They include the correction of baseline drift due to the accumulation of material on the SAXS capillary walls, and the Gaussian decomposition of non-baseline-resolved HPLC-SAXS elution peaks. It was thus possible to resolve at least two species co-eluting under the fibrinogen main monomer peak, probably resulting from in-column degradation, and two others under an oligomers peak. The overall and cross-sectional radii of gyration, molecular mass and mass/length ratio of all species were determined using the manual or semi-automated procedures available within the US-SOMO SAS module. Differences between monomeric species and linear and sideways oligomers were thus identified and rationalized. This new US-SOMO version additionally contains several computational and graphical tools, implementing functionalities such as the mapping of residues contributing to particular regions of P(r), and an advanced module for the comparison of primary I(q) versus q data with model curves computed from atomic level structures or bead models. It should be of great help in multi-resolution studies involving hydrodynamics, solution scattering and crystallographic/NMR data.
Keywords: HPLC-SAXS; bovine serum albumin; chromatography; fibrinogen; multi-resolution modeling; small-angle scattering.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials