

A perspective on proteomics in cell biology
- PMID: 24284280
- PMCID: PMC3989996
- DOI: 10.1016/j.tcb.2013.10.010
A perspective on proteomics in cell biology
Abstract
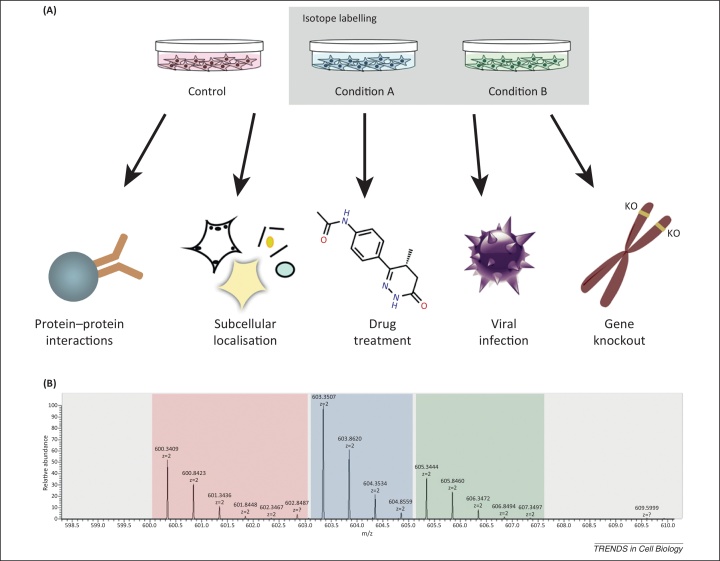
During the past 15 years mass spectrometry (MS)-based analyses have become established as the method of choice for direct protein identification and measurement. Owing to the remarkable improvements in the sensitivity and resolution of MS instruments, this technology has revolutionised the opportunities available for the system-wide characterisation of proteins, with wide applications across virtually the whole of cell biology. In this article we provide a perspective on the current state of the art and discuss how the future of cell biology research may benefit from further developments and applications in the field of MS and proteomics, highlighting the major challenges ahead for the community in organising the effective sharing and integration of the resulting data mountain.
Copyright © 2013 Elsevier Ltd. All rights reserved.
Figures



References
-
- Lamond A.I., Mann M. Cell biology and the genome projects a concerted strategy for characterizing multiprotein complexes by using mass spectrometry. Trends Cell Biol. 1997;7:139–142. - PubMed
-
- Eng J., et al. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. - PubMed
-
- Craig R., Beavis R.C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. - PubMed
-
- Perkins D.N., et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. - PubMed
-
- Cox J., et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
