

Mutational and fitness landscapes of an RNA virus revealed through population sequencing
- PMID: 24284629
- PMCID: PMC4111796
- DOI: 10.1038/nature12861
Mutational and fitness landscapes of an RNA virus revealed through population sequencing
Abstract
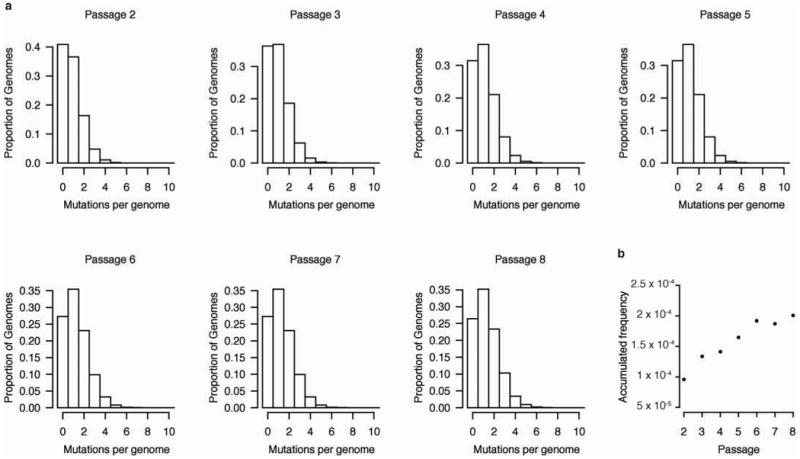
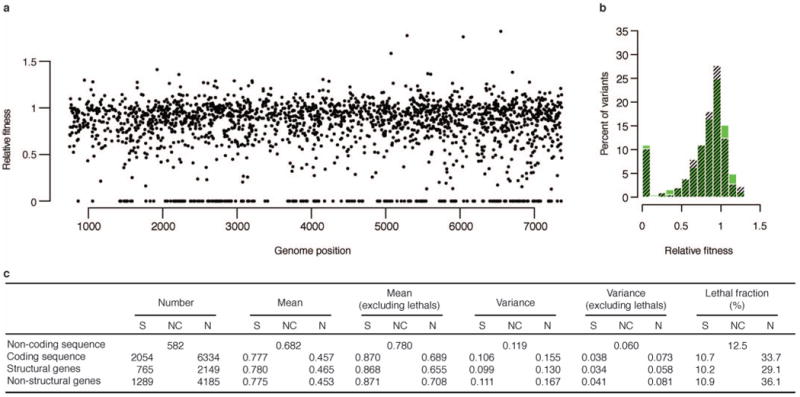
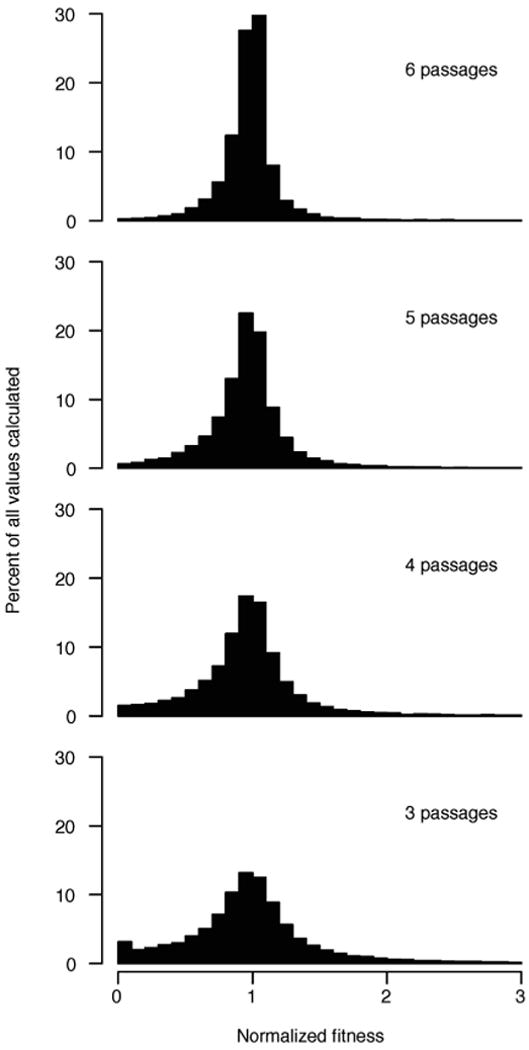
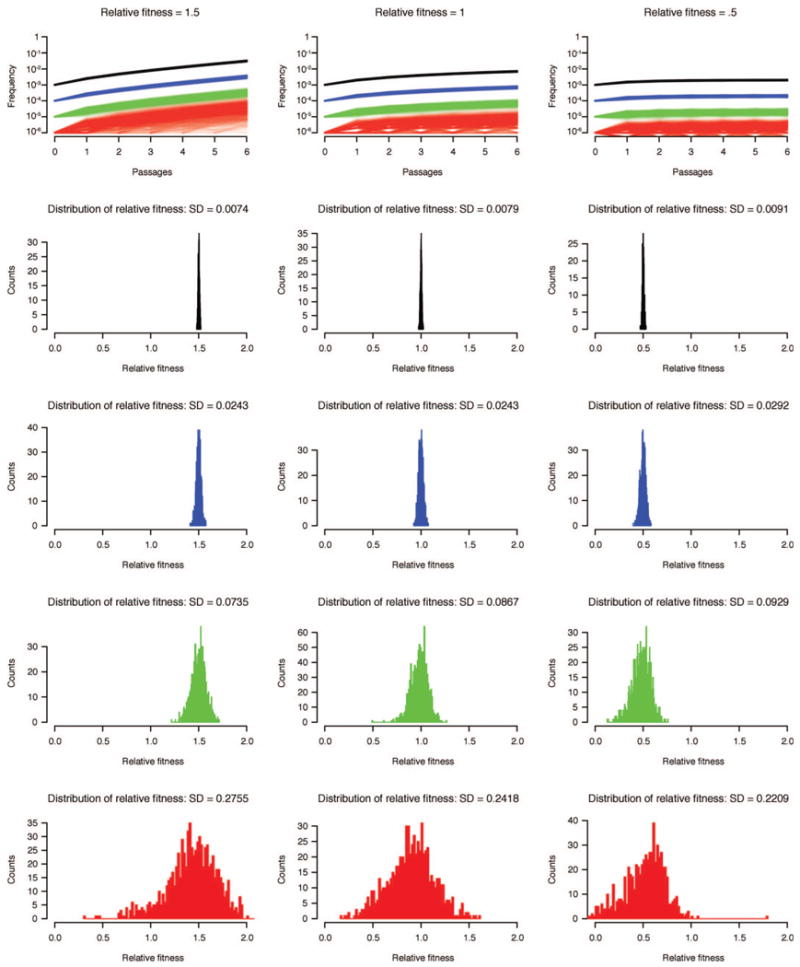
RNA viruses exist as genetically diverse populations. It is thought that diversity and genetic structure of viral populations determine the rapid adaptation observed in RNA viruses and hence their pathogenesis. However, our understanding of the mechanisms underlying virus evolution has been limited by the inability to accurately describe the genetic structure of virus populations. Next-generation sequencing technologies generate data of sufficient depth to characterize virus populations, but are limited in their utility because most variants are present at very low frequencies and are thus indistinguishable from next-generation sequencing errors. Here we present an approach that reduces next-generation sequencing errors and allows the description of virus populations with unprecedented accuracy. Using this approach, we define the mutation rates of poliovirus and uncover the mutation landscape of the population. Furthermore, by monitoring changes in variant frequencies on serially passaged populations, we determined fitness values for thousands of mutations across the viral genome. Mapping of these fitness values onto three-dimensional structures of viral proteins offers a powerful approach for exploring structure-function relationships and potentially uncovering new functions. To our knowledge, our study provides the first single-nucleotide fitness landscape of an evolving RNA virus and establishes a general experimental platform for studying the genetic changes underlying the evolution of virus populations.
Figures
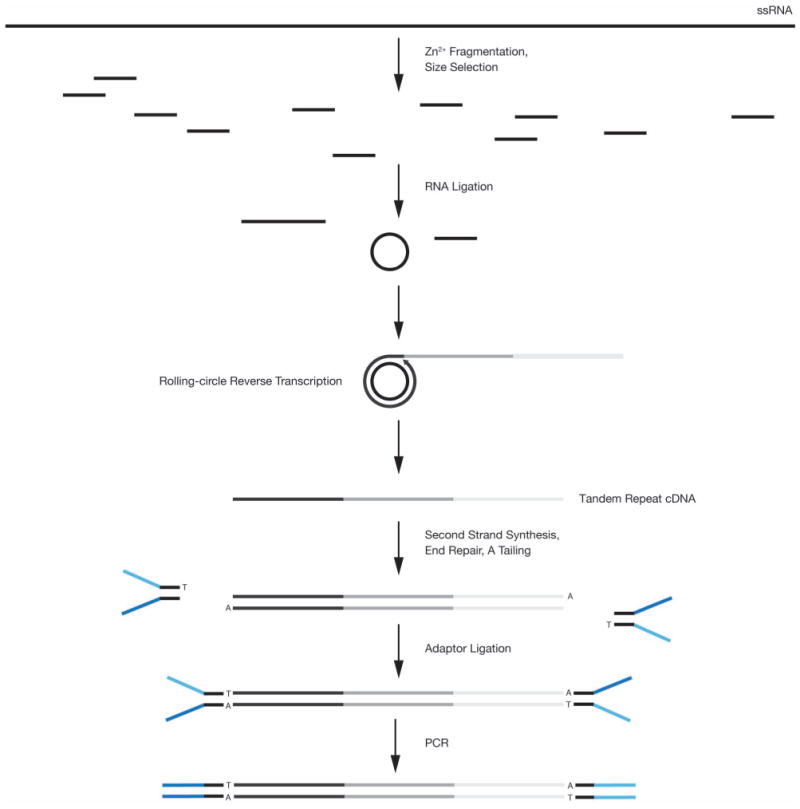
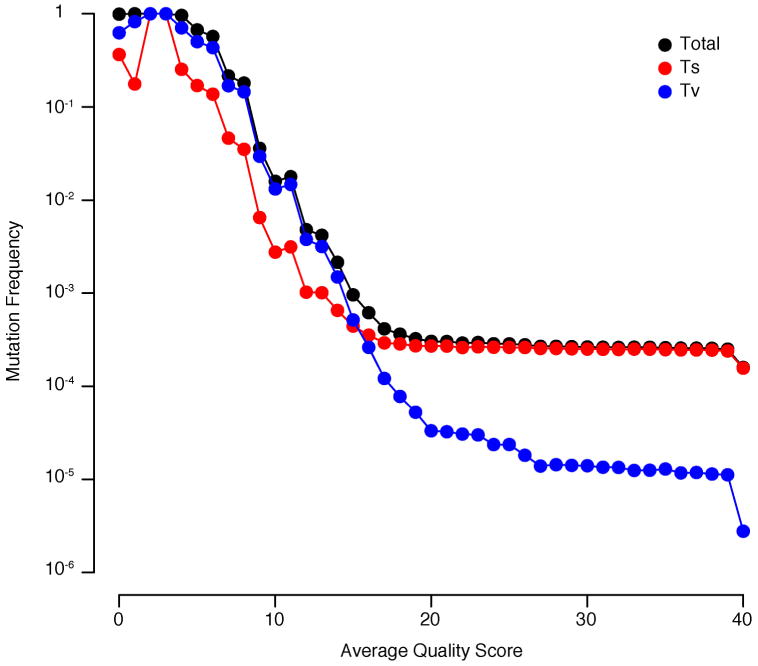
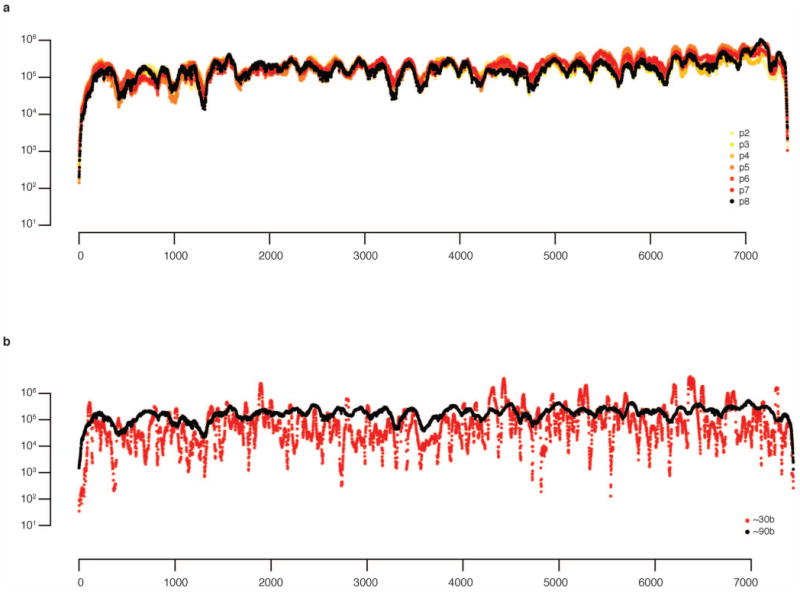
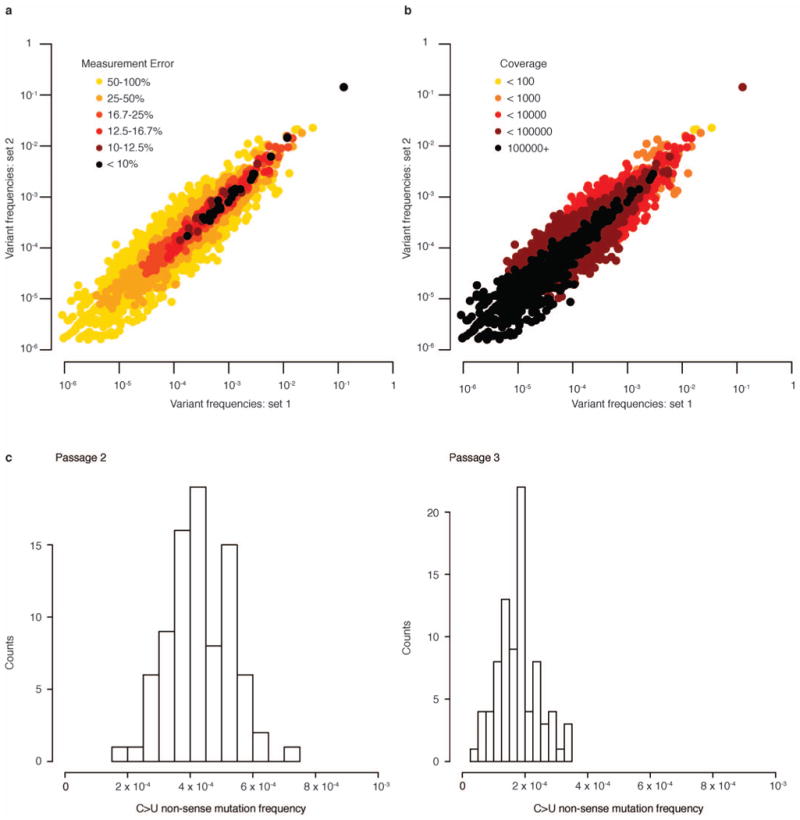
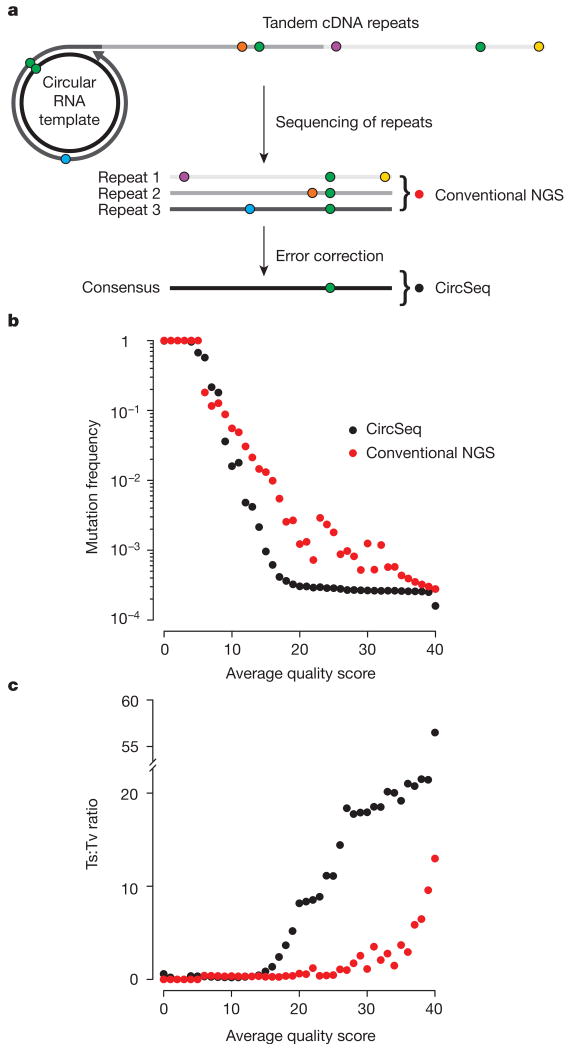
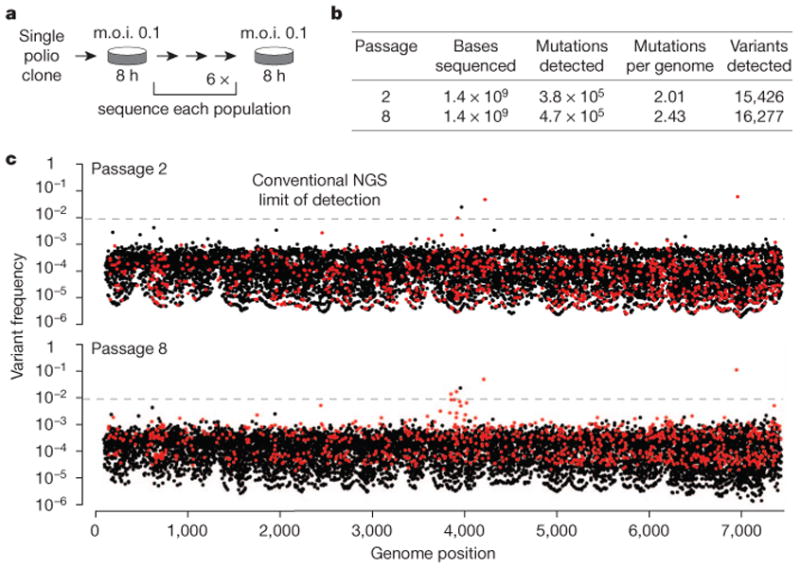
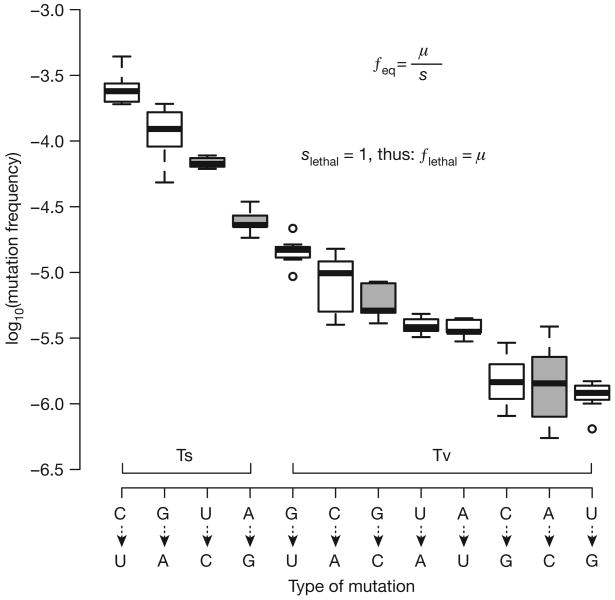
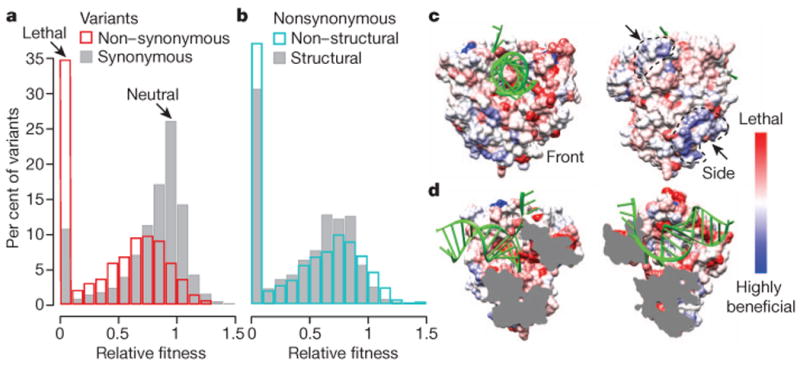
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
