

HIV-1 Vpr induces adipose dysfunction in vivo through reciprocal effects on PPAR/GR co-regulation
- PMID: 24285483
- PMCID: PMC4009012
- DOI: 10.1126/scitranslmed.3007148
HIV-1 Vpr induces adipose dysfunction in vivo through reciprocal effects on PPAR/GR co-regulation
Abstract
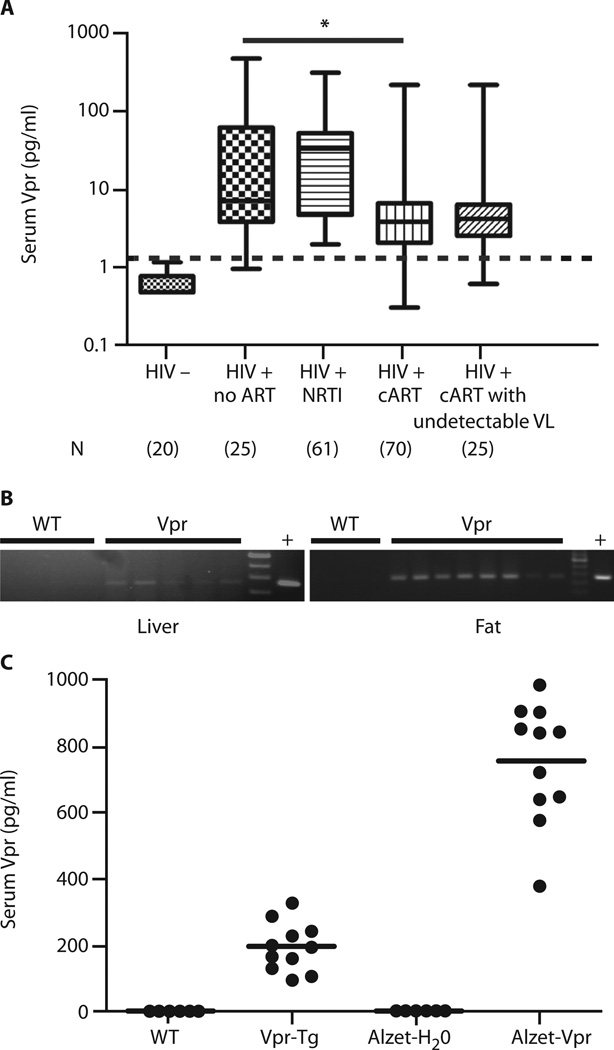
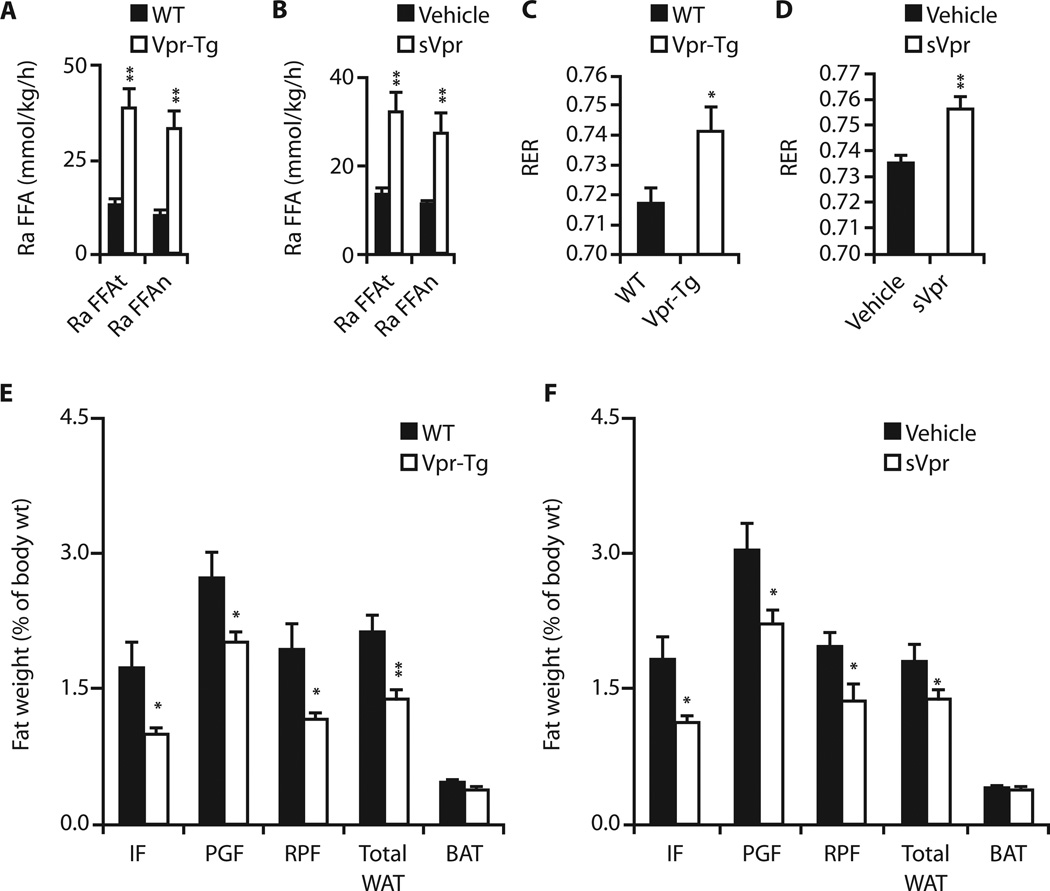
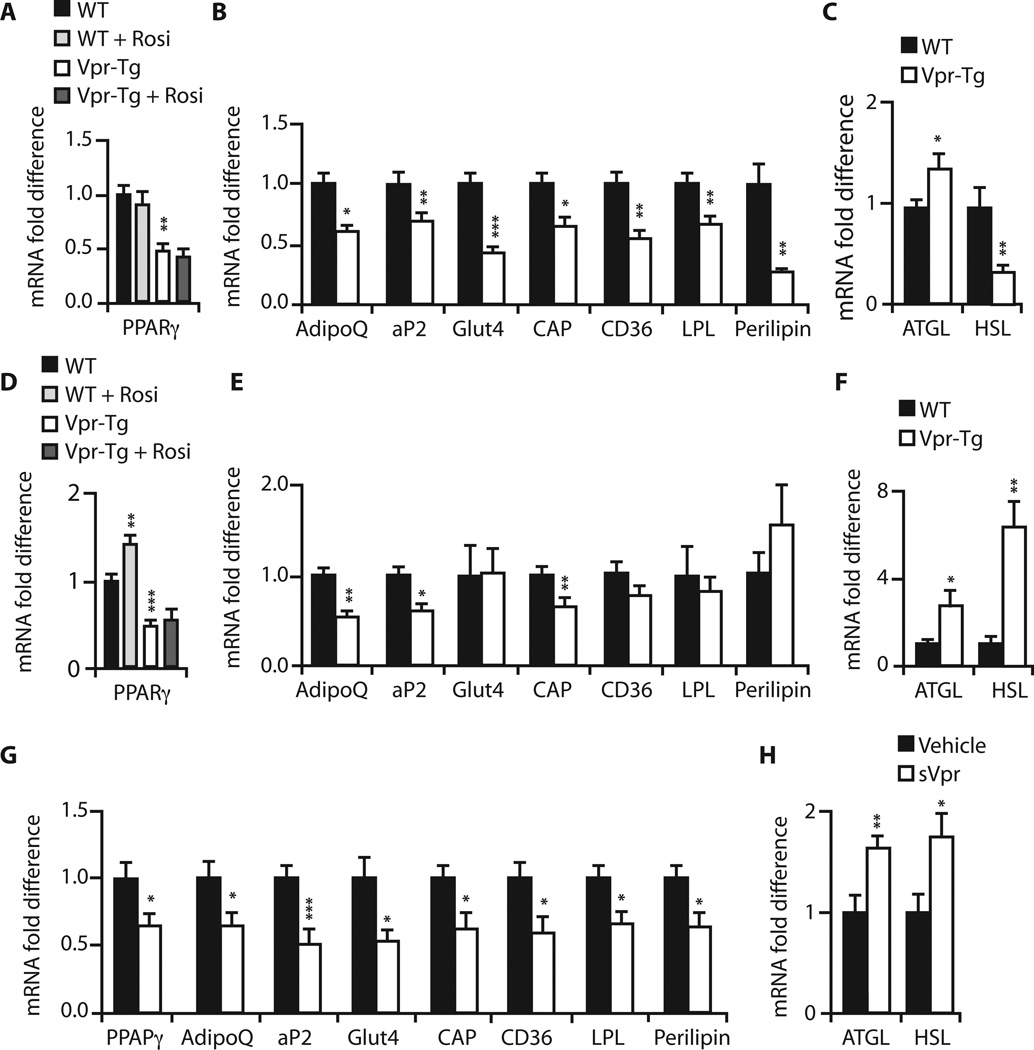
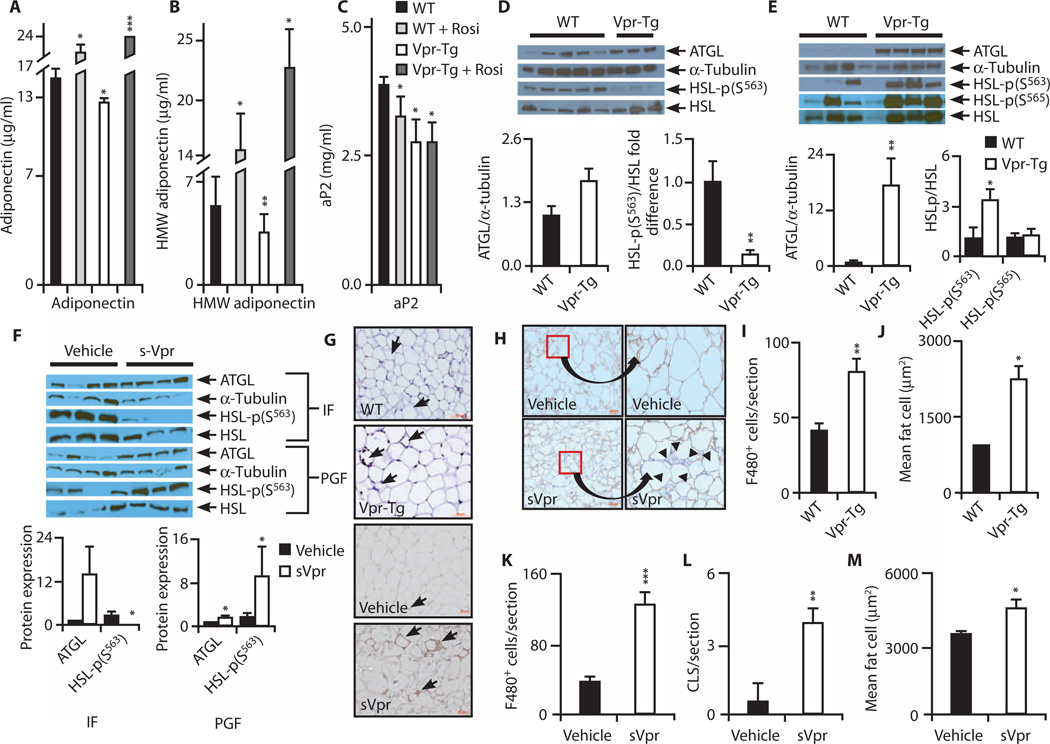
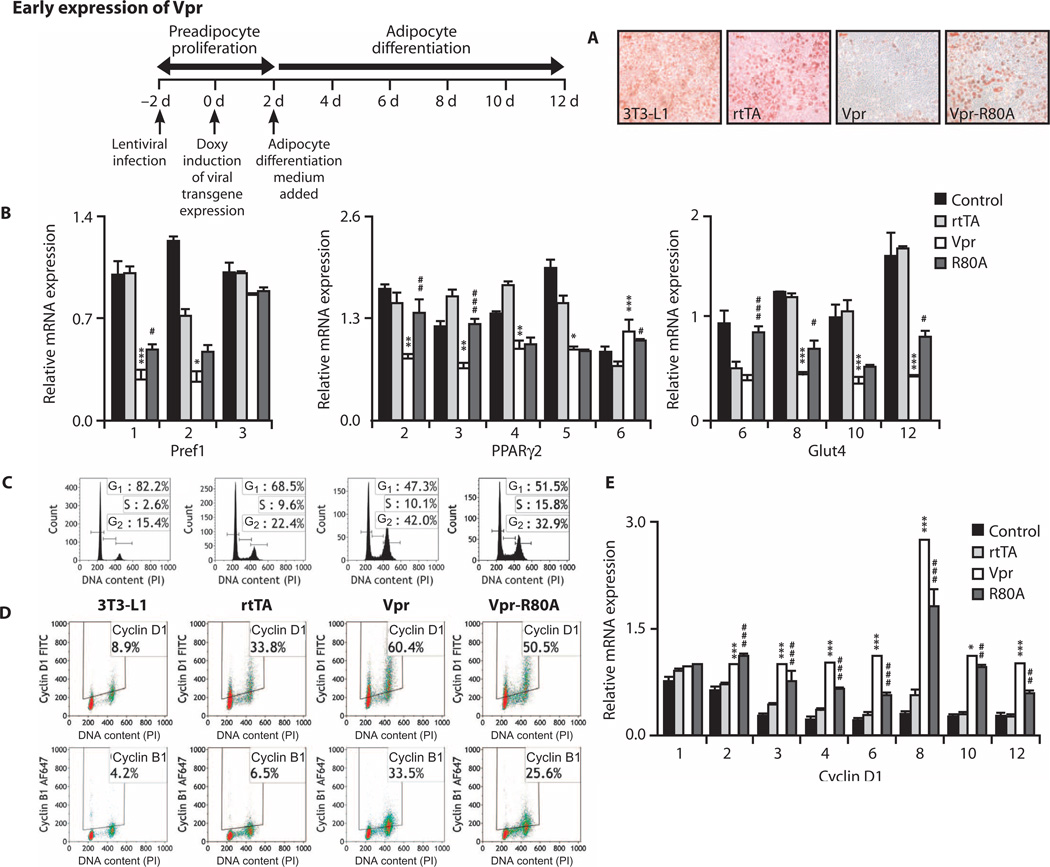
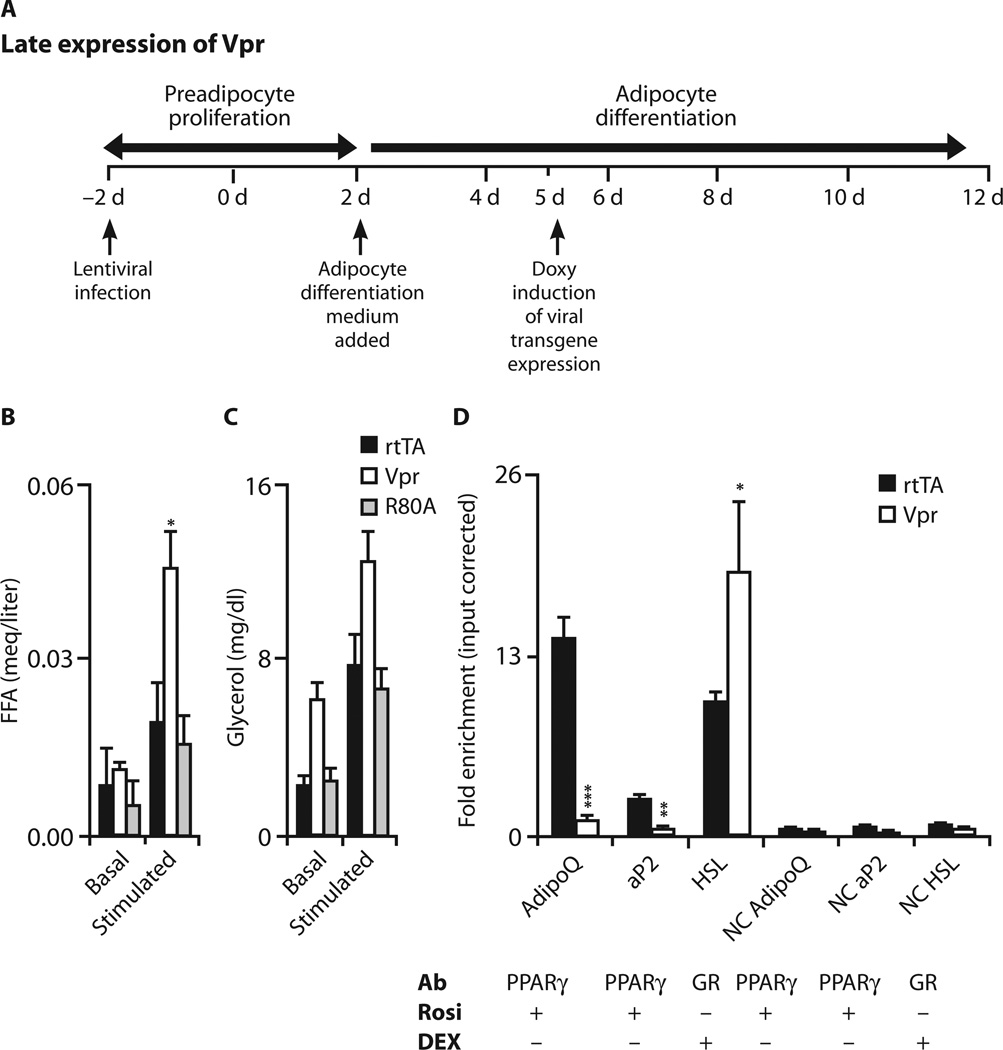
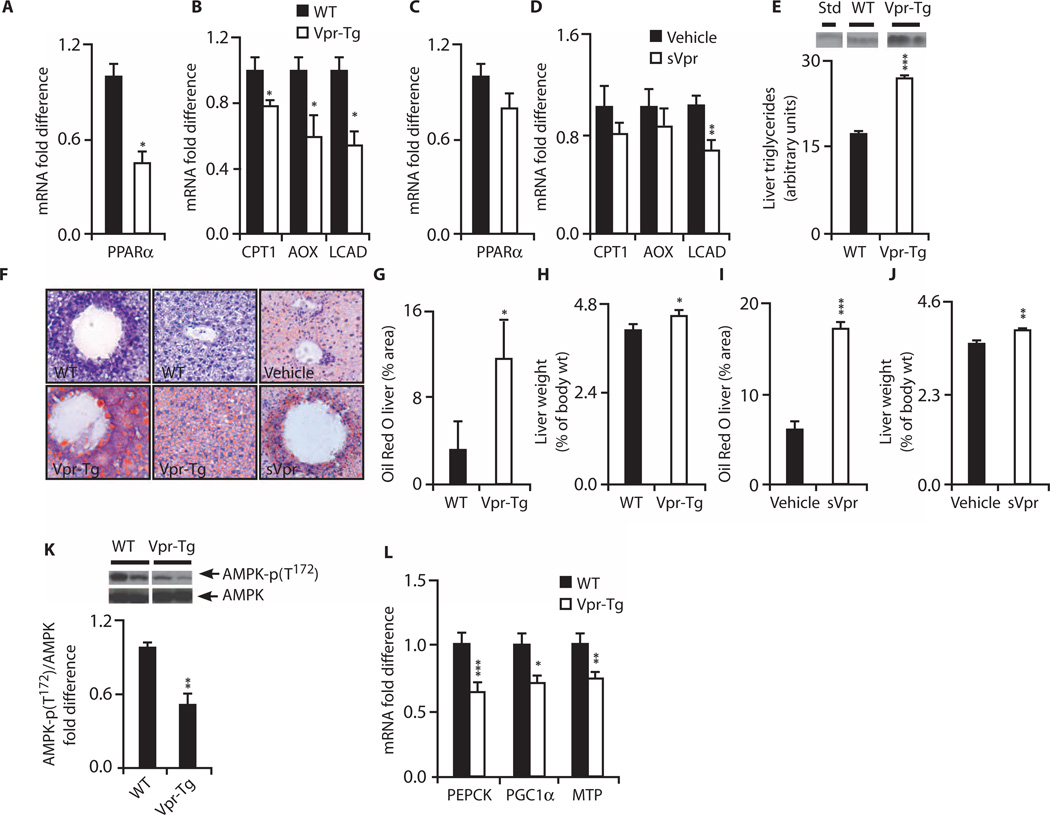
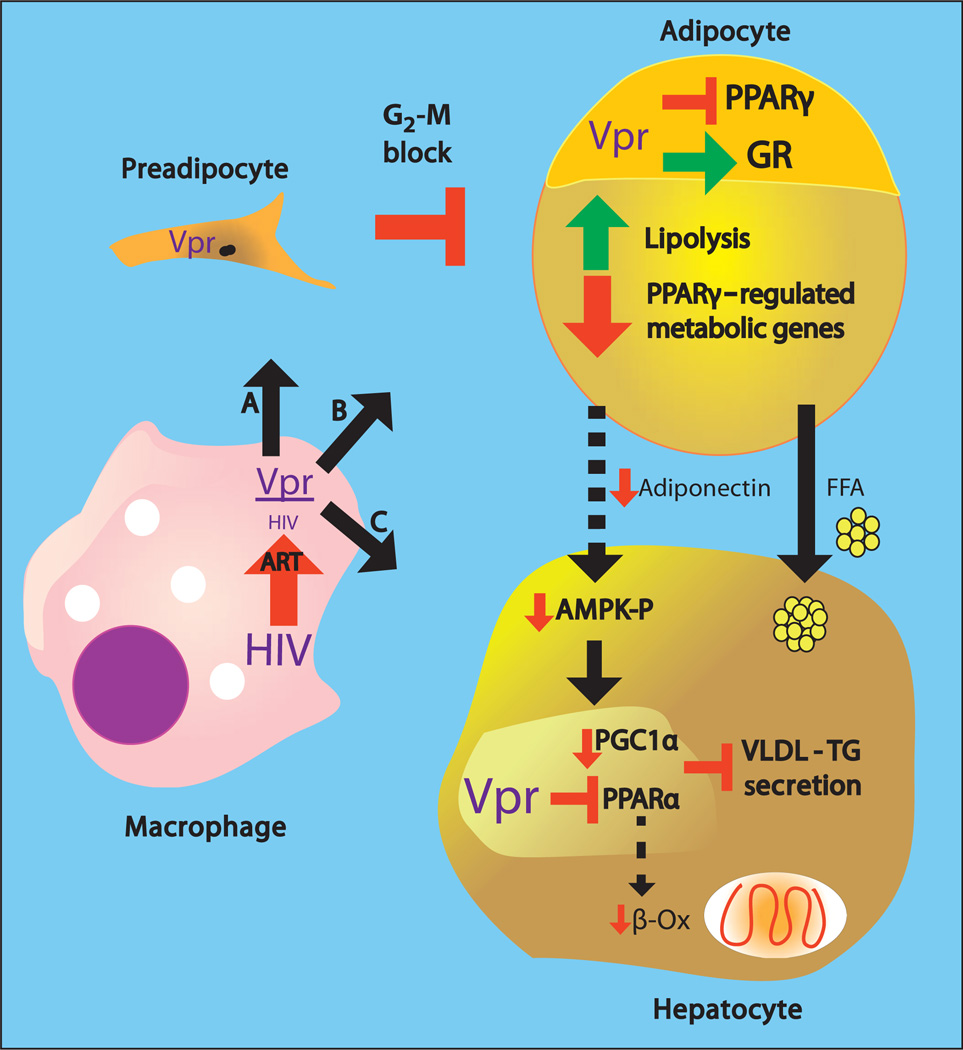
Viral infections, such as HIV, have been linked to obesity, but mechanistic evidence that they cause adipose dysfunction in vivo is lacking. We investigated a pathogenic role for the HIV-1 accessory protein viral protein R (Vpr), which can coactivate the glucocorticoid receptor (GR) and co-repress peroxisome proliferator-activated receptor γ (PPARγ) in vitro, in HIV-associated adipose dysfunction. Vpr circulated in the blood of most HIV-infected patients tested, including those on antiretroviral therapy (ART) with undetectable viral load. Vpr-mediated mechanisms were dissected in vivo using mouse models expressing the Vpr transgene in adipose tissues and liver (Vpr-Tg) or infused with synthetic Vpr. Both models demonstrated accelerated whole-body lipolysis, hyperglycemia and hypertriglyceridemia, and tissue-specific findings. Fat depots in these mice had diminished mass, macrophage infiltration, and blunted PPARγ target gene expression but increased GR target gene expression. In liver, we observed blunted PPARα target gene expression, steatosis with decreased adenosine monophosphate-activated protein kinase activity, and insulin resistance. Similar to human HIV-infected patients, Vpr circulated in the serum of Vpr-Tg mice. Vpr blocked differentiation in preadipocytes through cell cycle arrest, whereas in mature adipocytes, it increased lipolysis with reciprocally altered association of PPARγ and GR with their target promoters. These results delineate a distinct pathogenic sequence: Vpr, released from HIV-1 in tissue reservoirs after ART, can disrupt PPAR/GR co-regulation and cell cycle control to produce adipose dysfunction and hepatosteatosis. Confirmation of these mechanisms in HIV patients could lead to targeted treatment of the metabolic complications with Vpr inhibitors, GR antagonists, or PPARγ/PPARα agonists.
Figures
Similar articles
-
Human immunodeficiency virus (HIV)-1 viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor {gamma} and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy.Mol Endocrinol. 2008 Feb;22(2):234-47. doi: 10.1210/me.2007-0124. Epub 2007 Oct 11. Mol Endocrinol. 2008. PMID: 17932108 Free PMC article.
-
HIV-1 viral protein R (Vpr) induces fatty liver in mice via LXRα and PPARα dysregulation: implications for HIV-specific pathogenesis of NAFLD.Sci Rep. 2017 Oct 17;7(1):13362. doi: 10.1038/s41598-017-13835-w. Sci Rep. 2017. PMID: 29042644 Free PMC article.
-
Viral mechanisms of adipose dysfunction: lessons from HIV-1 Vpr.Adipocyte. 2014 Oct 30;4(1):55-9. doi: 10.4161/adip.29852. eCollection 2015 Jan-Mar. Adipocyte. 2014. PMID: 26167403 Free PMC article.
-
Human immunodeficiency virus type-1 accessory protein Vpr: a causative agent of the AIDS-related insulin resistance/lipodystrophy syndrome?Ann N Y Acad Sci. 2004 Jun;1024:153-67. doi: 10.1196/annals.1321.013. Ann N Y Acad Sci. 2004. PMID: 15265780 Review.
-
Partner molecules of accessory protein Vpr of the human immunodeficiency virus type 1.DNA Cell Biol. 2004 Apr;23(4):193-205. doi: 10.1089/104454904773819789. DNA Cell Biol. 2004. PMID: 15142377 Review.
Cited by
-
HIV-1 Viral Protein R Couples Metabolic Inflexibility With White Adipose Tissue Thermogenesis.Diabetes. 2021 Sep;70(9):2014-2025. doi: 10.2337/db20-0888. Epub 2021 Jul 7. Diabetes. 2021. PMID: 34233931 Free PMC article.
-
HIV and antiretroviral therapy-related fat alterations.Nat Rev Dis Primers. 2020 Jun 18;6(1):48. doi: 10.1038/s41572-020-0181-1. Nat Rev Dis Primers. 2020. PMID: 32555389 Review.
-
Vpr and Its Cellular Interaction Partners: R We There Yet?Cells. 2019 Oct 24;8(11):1310. doi: 10.3390/cells8111310. Cells. 2019. PMID: 31652959 Free PMC article. Review.
-
The Immune Response in Adipocytes and Their Susceptibility to Infection: A Possible Relationship with Infectobesity.Int J Mol Sci. 2022 May 31;23(11):6154. doi: 10.3390/ijms23116154. Int J Mol Sci. 2022. PMID: 35682832 Free PMC article. Review.
-
Adipose Tissue Is a Neglected Viral Reservoir and an Inflammatory Site during Chronic HIV and SIV Infection.PLoS Pathog. 2015 Sep 24;11(9):e1005153. doi: 10.1371/journal.ppat.1005153. eCollection 2015 Sep. PLoS Pathog. 2015. PMID: 26402858 Free PMC article.
References
-
- Lyons MJ, Faust IM, Hemmes RB, Buskirk DR, Hirsch J, Zabriskie JB. A virally induced obesity syndrome in mice. Science. 1982;216:82–85. - PubMed
-
- Ramesh S, Sanyal AJ. Hepatitis C and nonalcoholic fatty liver disease. Semin. Liver Dis. 2004;24:399–413. - PubMed
-
- Dhurandhar NV. A framework for identification of infections that contribute to human obesity. Lancet Infect. Dis. 2011;11:963–969. - PubMed
-
- Carr A, Miller J, Law M, Cooper DA. A syndrome of lipoatrophy, lactic acidaemia and liver dysfunction associated with HIV nucleoside analogue therapy: Contribution to protease inhibitor-related lipodystrophy syndrome. AIDS. 2000;14:F25–F32. - PubMed
-
- Pond CM. Long-term changes in adipose tissue in human disease. Proc. Nutr. Soc. 2001;60:365–374. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
