

Distinct roles for μ-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration
- PMID: 24285894
- PMCID: PMC3841454
- DOI: 10.1523/JNEUROSCI.3293-13.2013
Distinct roles for μ-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration
Abstract
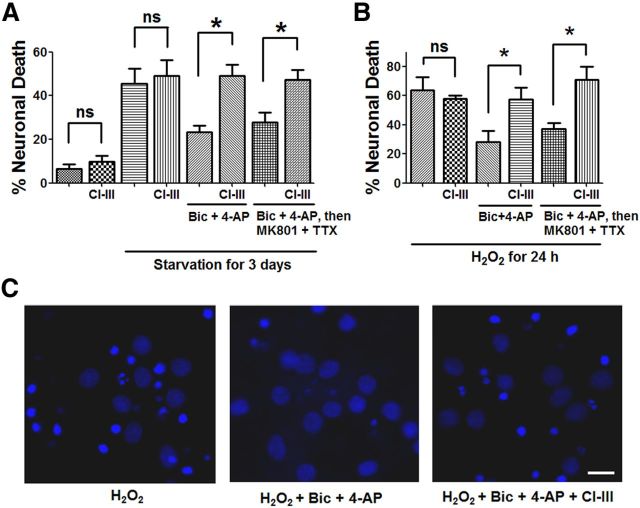
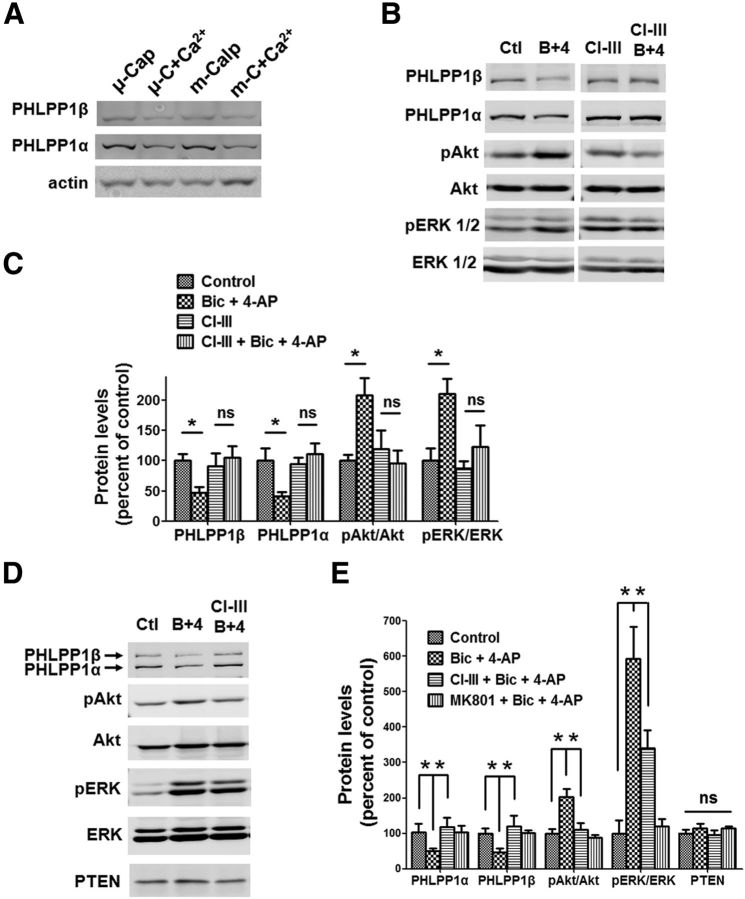
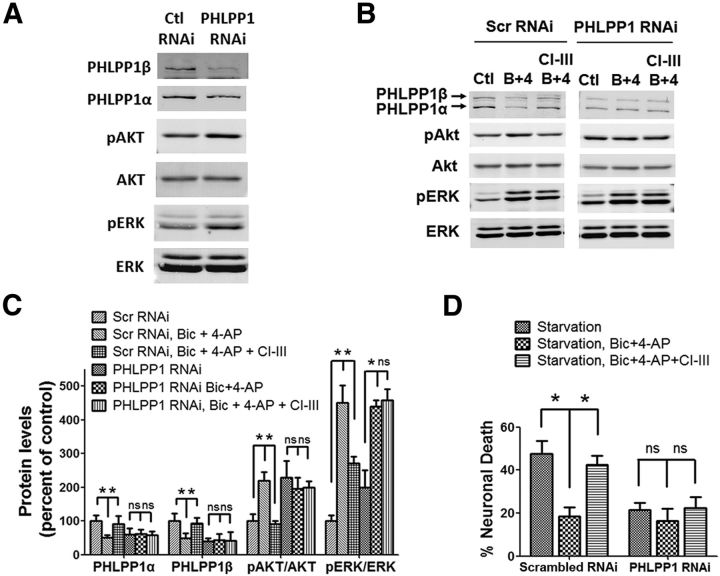
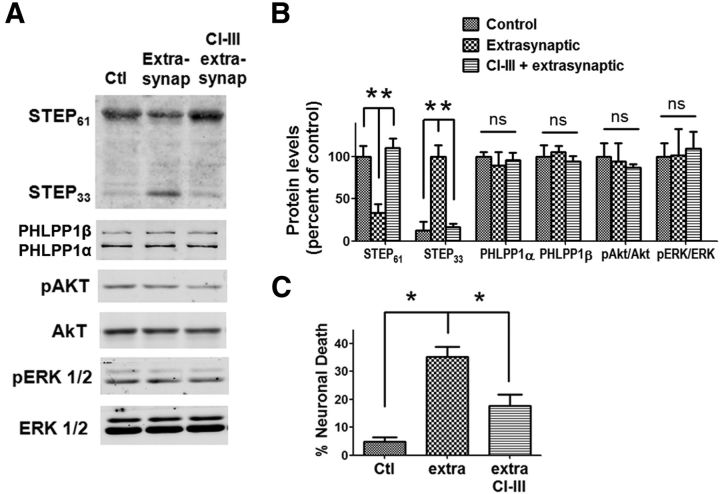
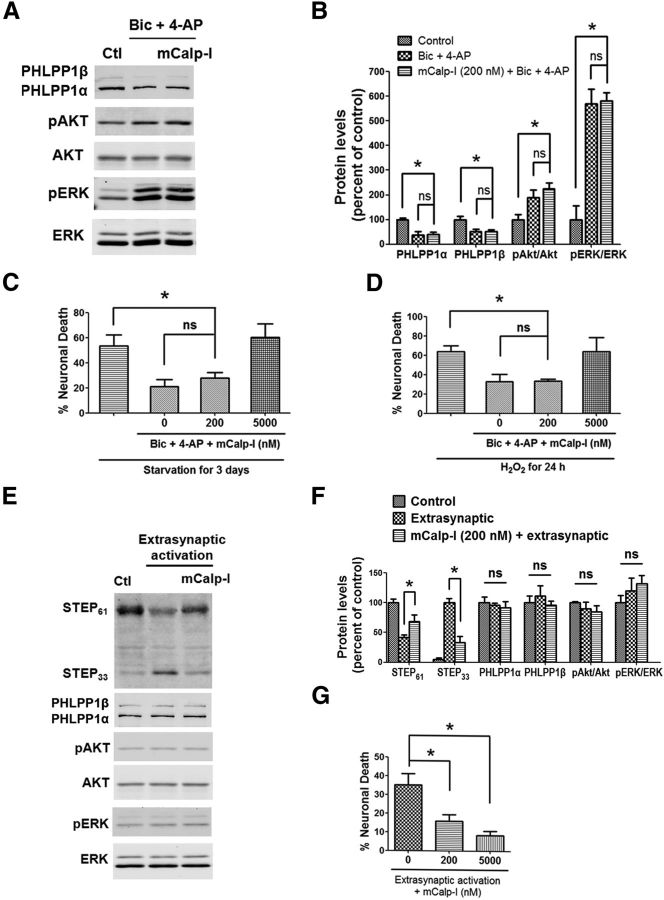
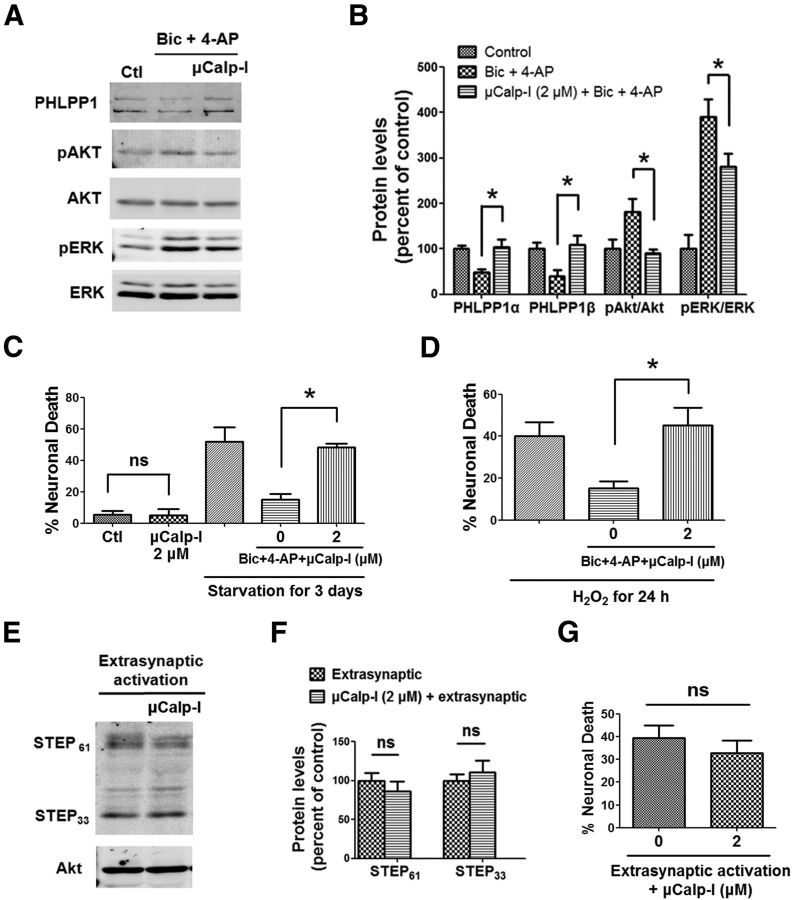
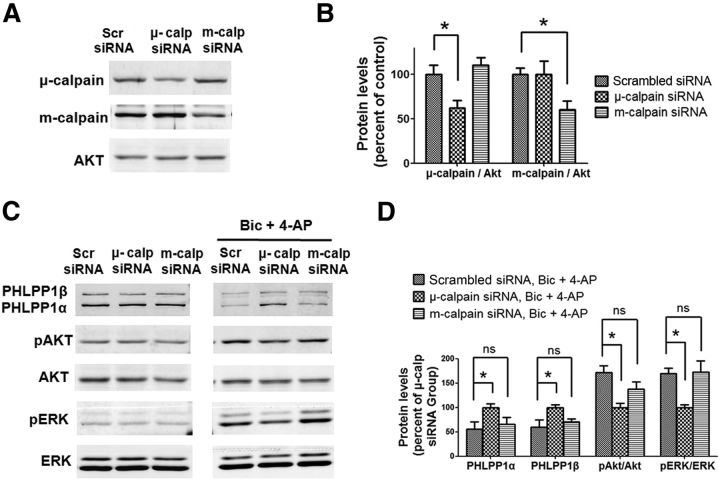
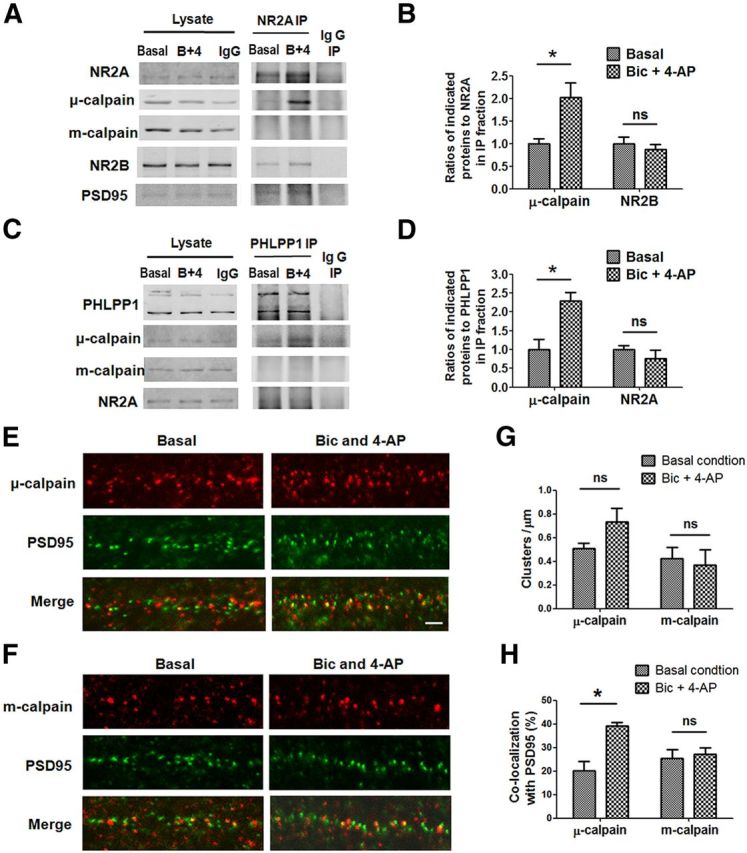
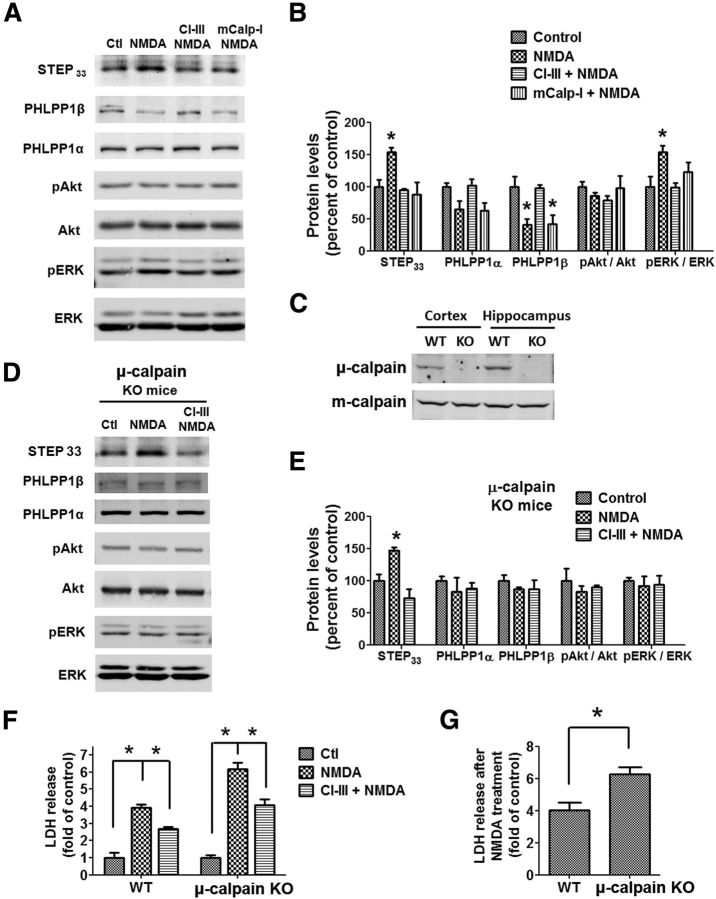
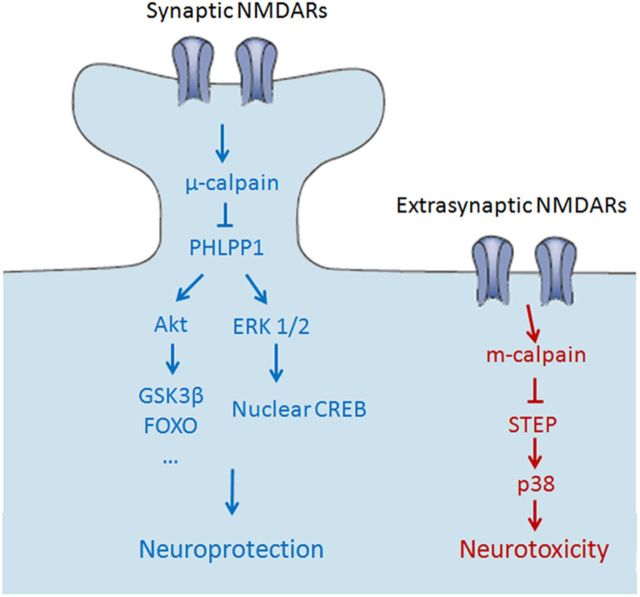
Prolonged calpain activation is widely recognized as a key component of neurodegeneration in a variety of pathological conditions. Numerous reports have also indicated that synaptic activation of NMDA receptors (NMDARs) provides neuroprotection against a variety of insults. Here, we report the paradoxical finding that such neuroprotection involves calpain activation. NMDAR activation in cultured rat cortical neurons was neuroprotective against starvation and oxidative stress-induced damage. It also resulted in the degradation of two splice variants of PH domain and Leucine-rich repeat Protein Phosphatase 1 (PHLPP1), PHLPP1α and PHLPP1β, which inhibit the Akt and ERK1/2 pathways. Synaptic NMDAR-induced neuroprotection and PHLPP1 degradation were blocked by calpain inhibition. Lentiviral knockdown of PHLPP1 mimicked the neuroprotective effects of synaptic NMDAR activation and occluded the effects of calpain inhibition on neuroprotection. In contrast to synaptic NMDAR activation, extrasynaptic NMDAR activation had no effect on PHLPP1 and the Akt and ERK1/2 pathways, but resulted in calpain-mediated degradation of striatal-enriched protein tyrosine phosphatase (STEP) and neuronal death. Using μ-calpain- and m-calpain-selective inhibitors and μ-calpain and m-calpain siRNAs, we found that μ-calpain-dependent PHLPP1 cleavage was involved in synaptic NMDAR-mediated neuroprotection, while m-calpain-mediated STEP degradation was associated with extrasynaptic NMDAR-induced neurotoxicity. Furthermore, m-calpain inhibition reduced while μ-calpain knockout exacerbated NMDA-induced neurotoxicity in acute mouse hippocampal slices. Thus, synaptic NMDAR-coupled μ-calpain activation is neuroprotective, while extrasynaptic NMDAR-coupled m-calpain activation is neurodegenerative. These results help to reconcile a number of contradictory results in the literature and have critical implications for the understanding and potential treatment of neurodegenerative diseases.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous