

Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy
- PMID: 24285972
- PMCID: PMC3840141
- DOI: 10.3988/jcn.2013.9.4.283
Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy
Abstract
Background: X-linked Charcot-Marie-Tooth disease type 5 (CMTX5) is caused by mutations in the gene encoding phosphoribosyl pyrophosphate synthetase I (PRPS1). There has been only one case report of CMTX5 patients. The aim of this study was to identify the causative gene in a family with CMTX with peripheral neuropathy and deafness.
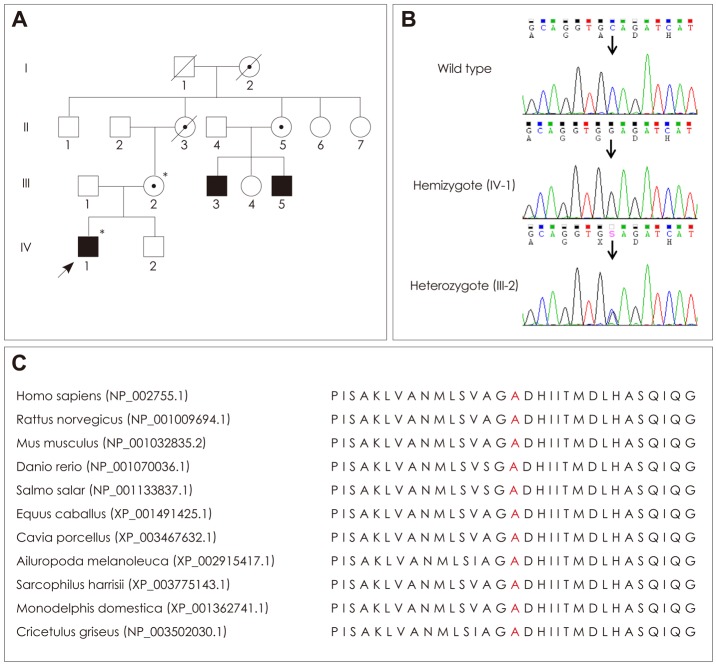
Case report: A Korean family with X-linked recessive CMT was enrolled. The age at the onset of hearing loss of the male proband was 5 months, and that of steppage gait was 6 years; he underwent cochlear surgery at the age of 12 years. In contrast to what was reported for the first patients with CMTX5, this patient did not exhibit optic atrophy. Furthermore, there was no cognitive impairment, respiratory dysfunction, or visual disturbance. Assessment of his family history revealed two male relatives with very similar clinical manifestations. Electrophysiological evaluations disclosed sensorineural hearing loss and peripheral neuropathy. Whole-exome sequencing identified a novel p.Ala121Gly (c.362C>G) PRPS1 mutation as the underlying genetic cause of the clinical phenotype.
Conclusions: A novel mutation of PRPS1 was identified in a CMTX5 family in which the proband had a phenotype of peripheral neuropathy with early-onset hearing loss, but no optic atrophy. The findings of this study will expand the clinical spectrum of X-linked recessive CMT and will be useful for the molecular diagnosis of clinically heterogeneous peripheral neuropathies.
Keywords: Charcot-Marie-Tooth disease type X5; deafness; exome; mutation; phosphoribosyl pyrophosphate synthetase I gene.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
References
-
- Kim HJ, Sohn KM, Shy ME, Krajewski KM, Hwang M, Park JH, et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5) Am J Hum Genet. 2007;81:552–558. - PMC - PubMed
-
- Rosenberg RN, Chutorian A. Familial opticoacoustic nerve degeneration and polyneuropathy. Neurology. 1967;17:827–832. - PubMed
-
- Sperling O, Eilam G, Sara-Persky-Brosh, De Vries A. Accelerated erythrocyte 5-phosphoribosyl-1-pyrophosphate synthesis. A familial abnormality associated with excessive uric acid production and gout. Biochem Med. 1972;6:310–316. - PubMed
-
- Arts WF, Loonen MC, Sengers RC, Slooff JL. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann Neurol. 1993;33:535–539. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
