

Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease
- PMID: 24290752
- PMCID: PMC3957429
- DOI: 10.1016/j.celrep.2013.10.042
Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease
Abstract
Autophagy dysfunction has been implicated in misfolded protein accumulation and cellular toxicity in several diseases. Whether alterations in autophagy also contribute to the pathology of lipid-storage disorders is not clear. Here, we show defective autophagy in Niemann-Pick type C1 (NPC1) disease associated with cholesterol accumulation, where the maturation of autophagosomes is impaired because of defective amphisome formation caused by failure in SNARE machinery, whereas the lysosomal proteolytic function remains unaffected. Expression of functional NPC1 protein rescues this defect. Inhibition of autophagy also causes cholesterol accumulation. Compromised autophagy was seen in disease-affected organs of Npc1 mutant mice. Of potential therapeutic relevance is that HP-β-cyclodextrin, which is used for cholesterol-depletion treatment, impedes autophagy, whereas stimulating autophagy restores its function independent of amphisome formation. Our data suggest that a low dose of HP-β-cyclodextrin that does not perturb autophagy, coupled with an autophagy inducer, may provide a rational treatment strategy for NPC1 disease.
Copyright © 2013 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
R.J. is an advisor to Stemgent and Fate Therapeutics.
Figures




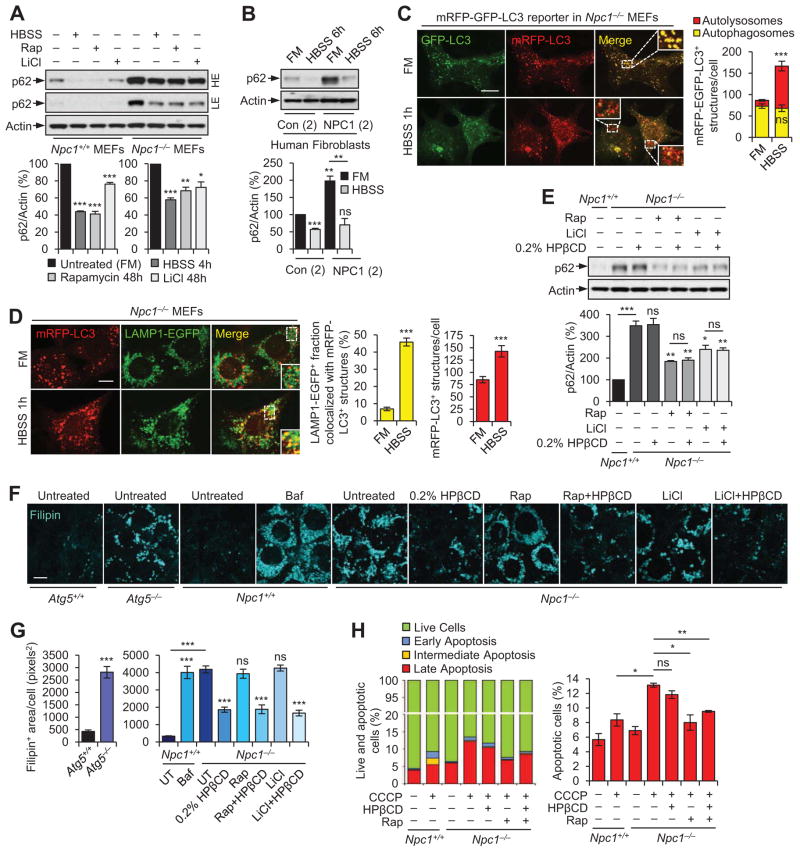


References
-
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–21892. - PubMed
-
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. - PubMed
-
- Fader CM, Sanchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta. 2009;1793:1901–1916. - PubMed
-
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
