

Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver
- PMID: 24294945
- PMCID: PMC4123468
- DOI: 10.1089/ars.2013.5486
Reactive oxygen and nitrogen species in steatotic hepatocytes: a molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver
Abstract
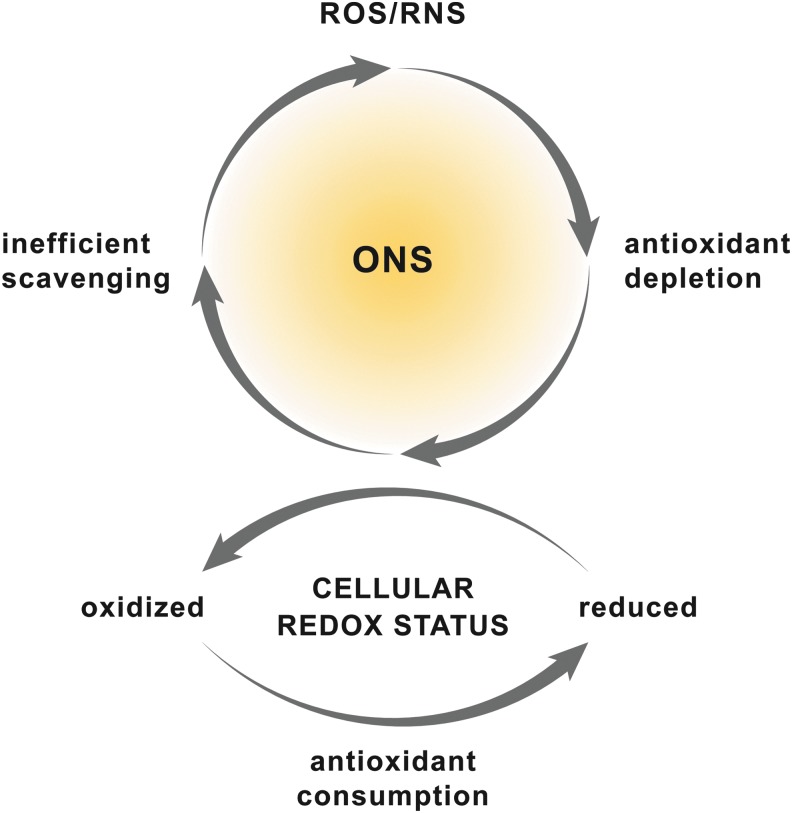
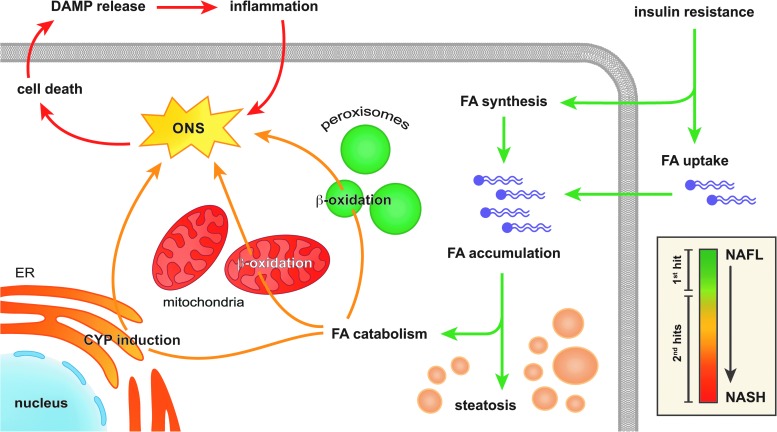
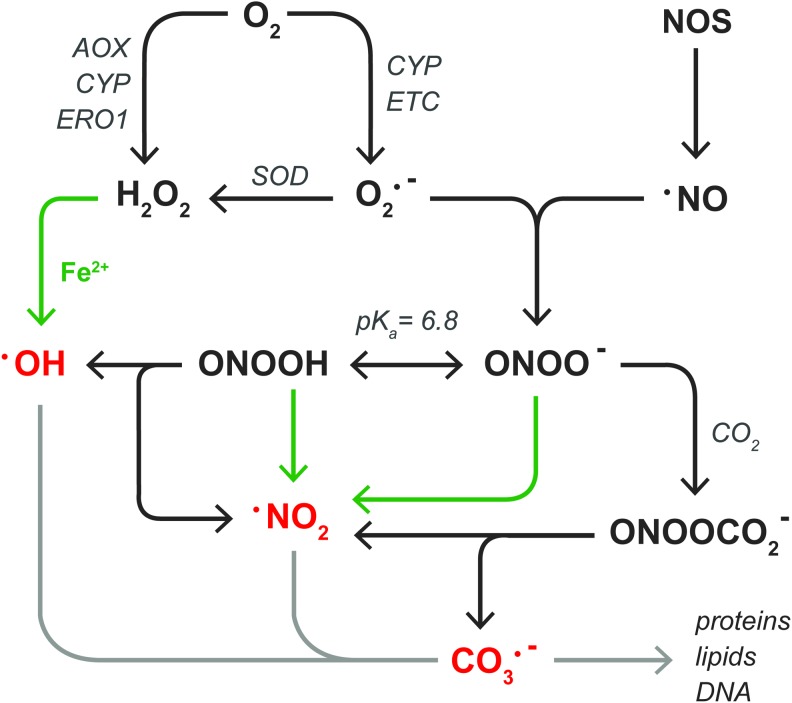
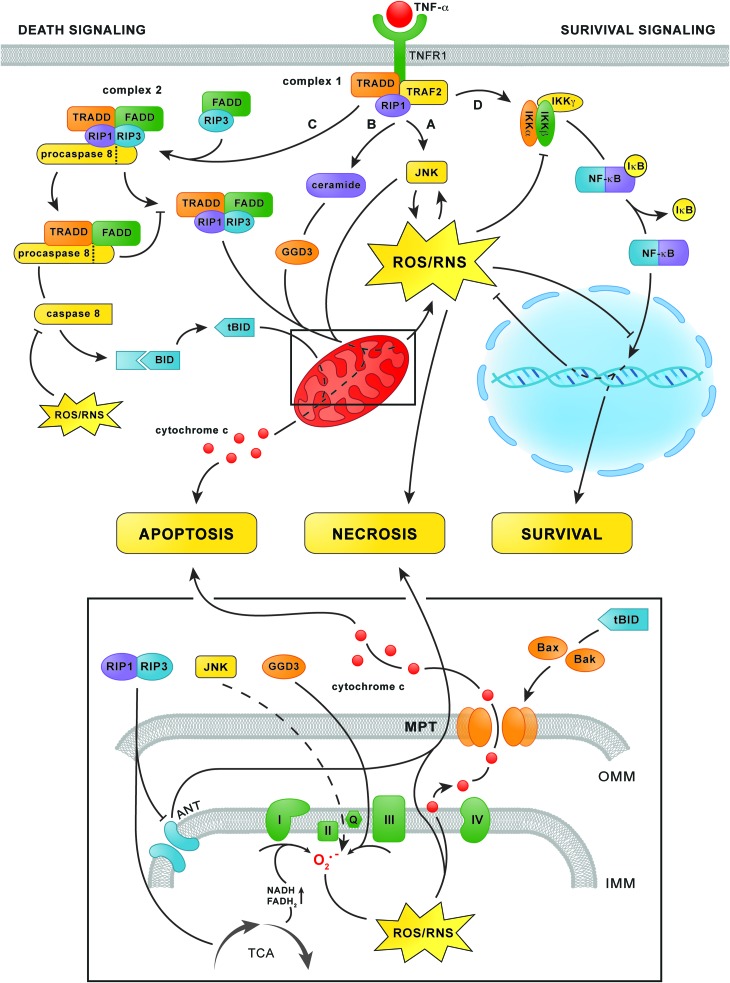
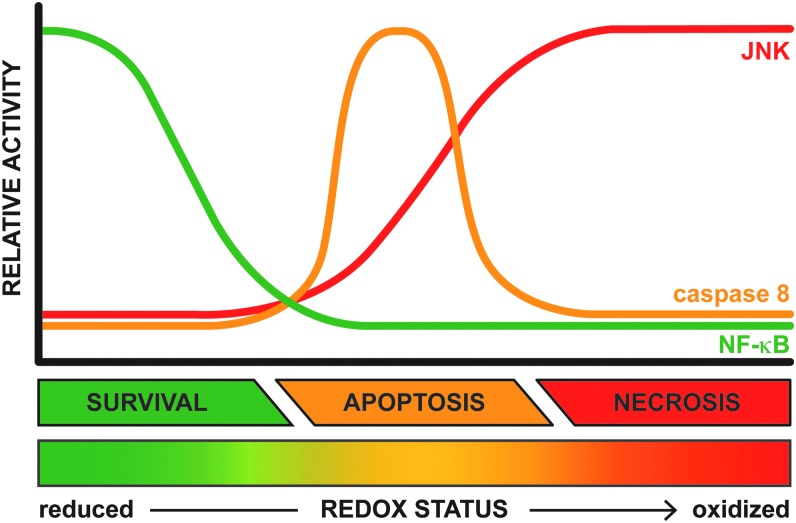
Significance: Hepatic ischemia-reperfusion (IR) injury results from the temporary deprivation of hepatic blood supply and is a common side effect of major liver surgery (i.e., transplantation or resection). IR injury, which in most severe cases culminates in acute liver failure, is particularly pronounced in livers that are affected by non-alcoholic fatty liver disease (NAFLD). In NAFLD, fat-laden hepatocytes are damaged by chronic oxidative/nitrosative stress (ONS), a state that is acutely exacerbated during IR, leading to extensive parenchymal damage.
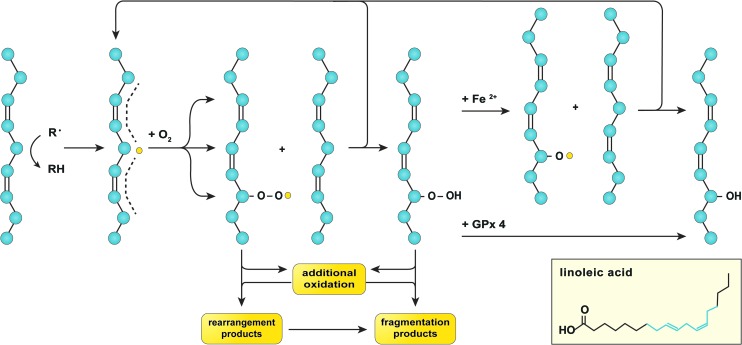
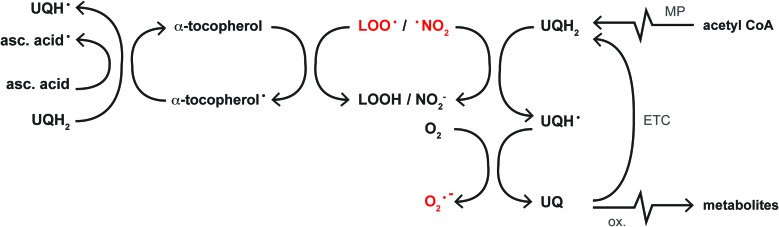
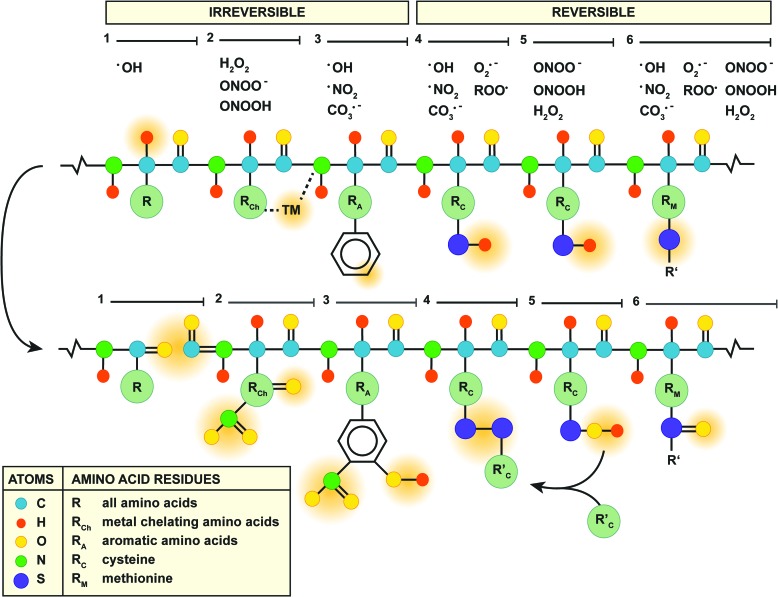
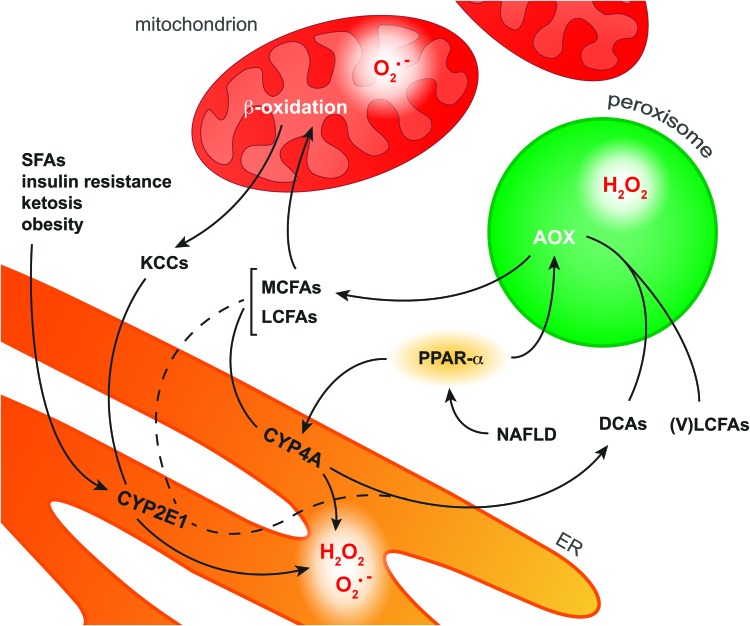
Recent advances: NAFLD triggers ONS via increased (extra)mitochondrial fatty acid oxidation and activation of the unfolded protein response. ONS is associated with widespread protein and lipid (per)oxidation, which reduces the hepatic antioxidative capacity and shifts the intracellular redox status toward an oxidized state. Moreover, activation of the transcription factor peroxisome proliferator-activated receptor α induces expression of mitochondrial uncoupling protein 2, resulting in depletion of cellular energy (ATP) reserves. The reduction in intracellular antioxidants and ATP in fatty livers consequently gives rise to severe ONS and necrotic cell death during IR.
Critical issues: Despite the fact that ONS mediates both NAFLD and IR injury, the interplay between the two conditions has never been described in detail. An integrative overview of the pathophysiology of NAFLD that renders steatotic hepatocytes more vulnerable to IR injury is therefore presented in the context of ONS.
Future directions: Effective methods should be devised to alleviate ONS and the consequences thereof in NAFLD before surgery in order to improve resilience of fatty livers to IR injury.
Figures
References
-
- Afri M, Gottlieb HE, and Frimer AA. Superoxide organic chemistry within the liposomal bilayer, part II: a correlation between location and chemistry. Free Radic Biol Med 32: 605–618, 2002 - PubMed
-
- Aguirre V, Uchida T, Yenush L, Davis R, and White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275: 9047–9054, 2000 - PubMed
-
- Allard JP, Aghdassi E, Mohammed S, Raman M, Avand G, Arendt BM, Jalali P, Kandasamy T, Prayitno N, Sherman M, Guindi M, Ma DW, and Heathcote JE. Nutritional assessment and hepatic fatty acid composition in non-alcoholic fatty liver disease (NAFLD): a cross-sectional study. J Hepatol 48: 300–307, 2008 - PubMed
-
- Alvarez B, Demicheli V, Durán R, Trujillo M, Cerveñansky C, Freeman BA, and Radi R. Inactivation of human Cu,Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic Biol Med 37: 813–822, 2004 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources