

Systems genetics approaches to understand complex traits
- PMID: 24296534
- PMCID: PMC3934510
- DOI: 10.1038/nrg3575
Systems genetics approaches to understand complex traits
Abstract
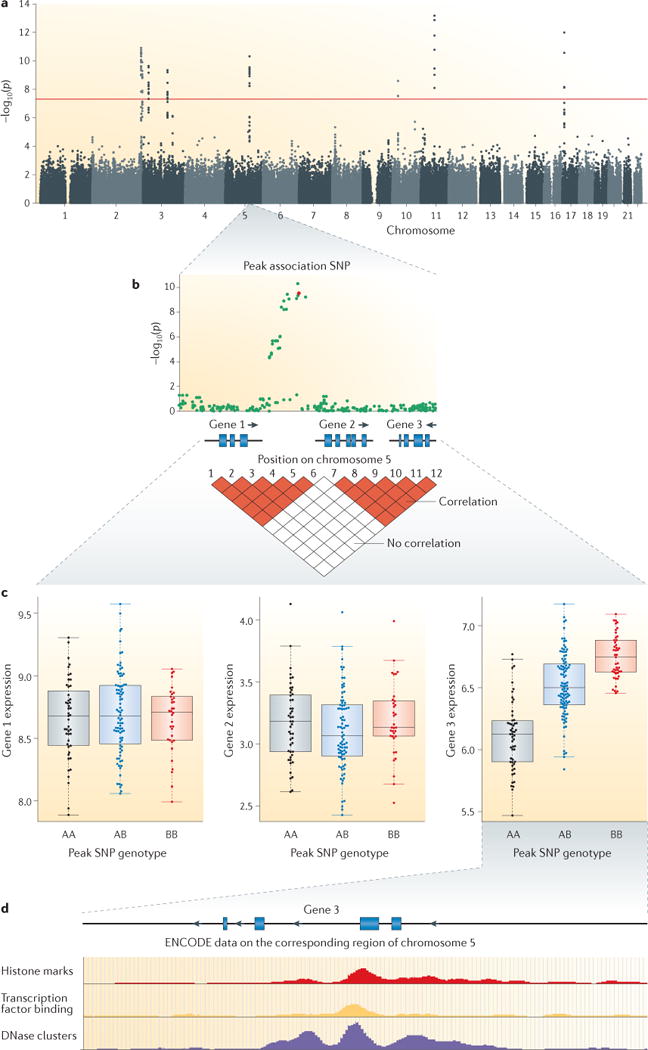
Systems genetics is an approach to understand the flow of biological information that underlies complex traits. It uses a range of experimental and statistical methods to quantitate and integrate intermediate phenotypes, such as transcript, protein or metabolite levels, in populations that vary for traits of interest. Systems genetics studies have provided the first global view of the molecular architecture of complex traits and are useful for the identification of genes, pathways and networks that underlie common human diseases. Given the urgent need to understand how the thousands of loci that have been identified in genome-wide association studies contribute to disease susceptibility, systems genetics is likely to become an increasingly important approach to understanding both biology and disease.
Conflict of interest statement
The authors declare no competing interests.
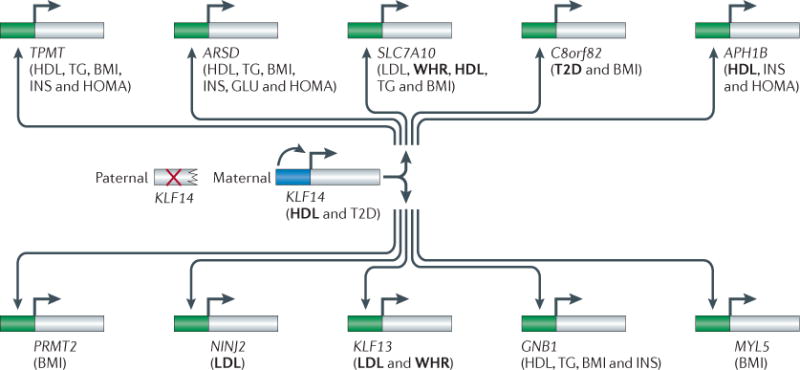
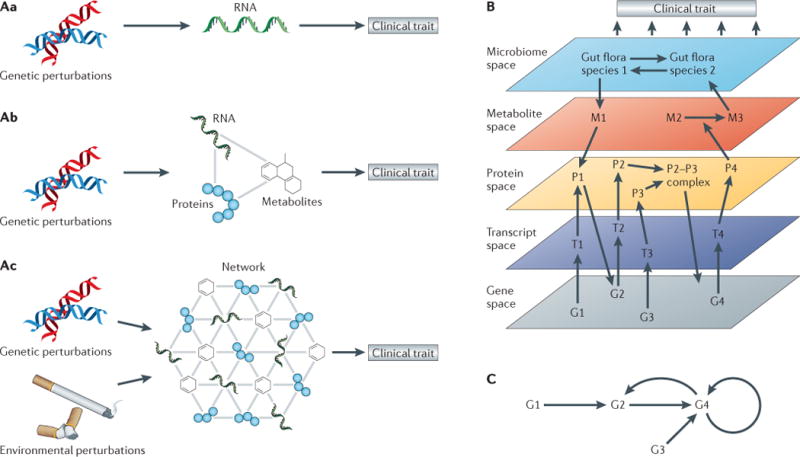
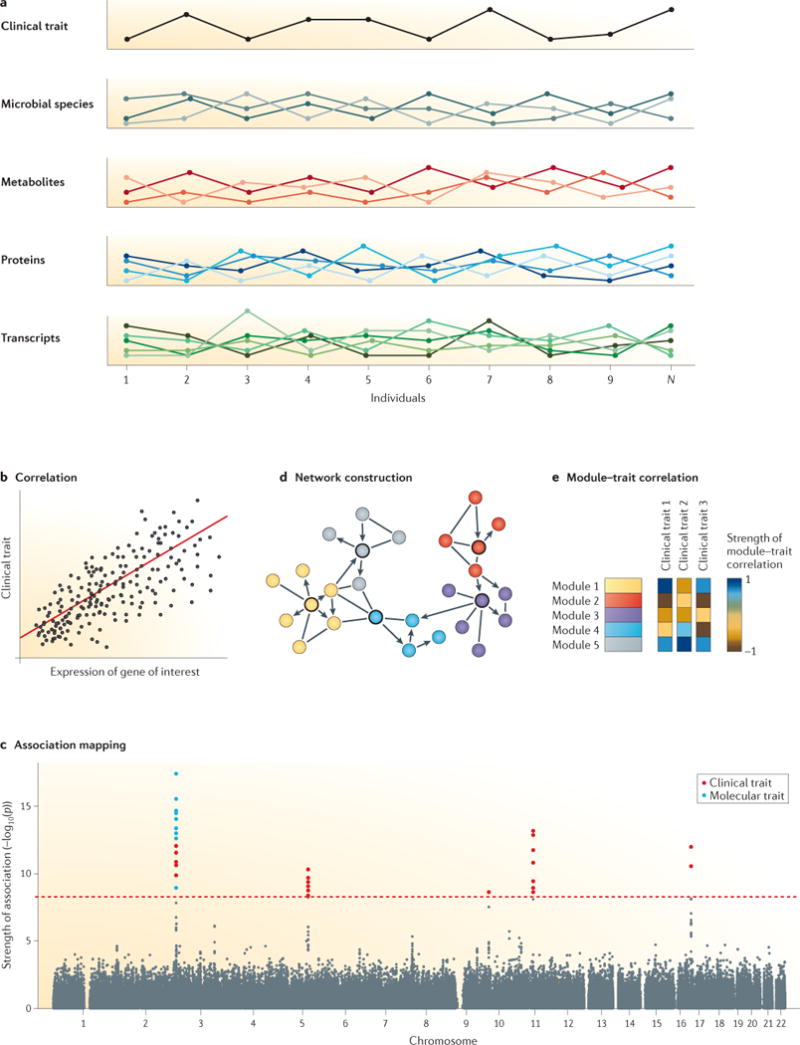
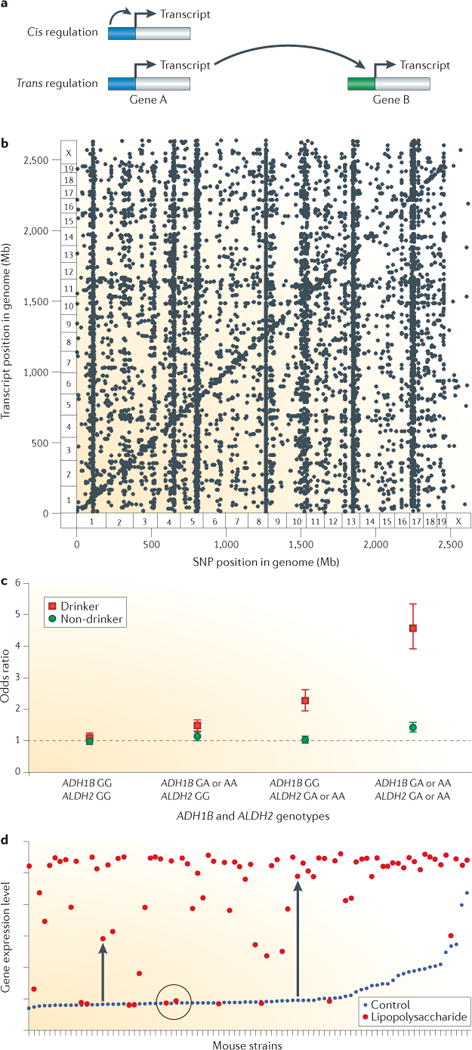
Figures
References
-
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4. Longman; 1996.
-
- Lynch M, Walsh JB. Genetics and Analysis of Quantitative Traits. Sinauer Associates; 1998.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
