

Host-directed therapeutics for tuberculosis: can we harness the host?
- PMID: 24296574
- PMCID: PMC3973381
- DOI: 10.1128/MMBR.00032-13
Host-directed therapeutics for tuberculosis: can we harness the host?
Abstract
Treatment of tuberculosis (TB) remains challenging, with lengthy treatment durations and complex drug regimens that are toxic and difficult to administer. Similar to the vast majority of antibiotics, drugs for Mycobacterium tuberculosis are directed against microbial targets. Although more effective drugs that target the bacterium may lead to faster cure of patients, it is possible that a biological limit will be reached that can be overcome only by adopting a fundamentally new treatment approach. TB regimens might be improved by including agents that target host pathways. Recent work on host-pathogen interactions, host immunity, and host-directed interventions suggests that supplementing anti-TB therapy with host modulators may lead to shorter treatment times, a reduction in lung damage caused by the disease, and a lower risk of relapse or reinfection. We undertook this review to identify molecular pathways of the host that may be amenable to modulation by small molecules for the treatment of TB. Although several approaches to augmenting standard TB treatment have been proposed, only a few have been explored in detail or advanced to preclinical and clinical studies. Our review focuses on molecular targets and inhibitory small molecules that function within the macrophage or other myeloid cells, on host inflammatory pathways, or at the level of TB-induced lung pathology.
Figures
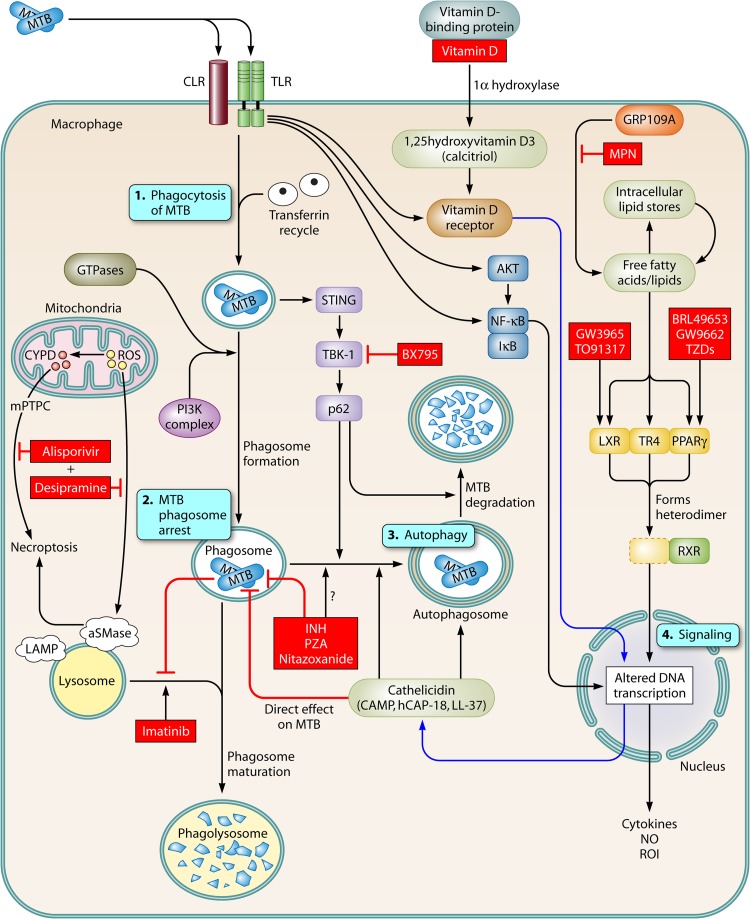
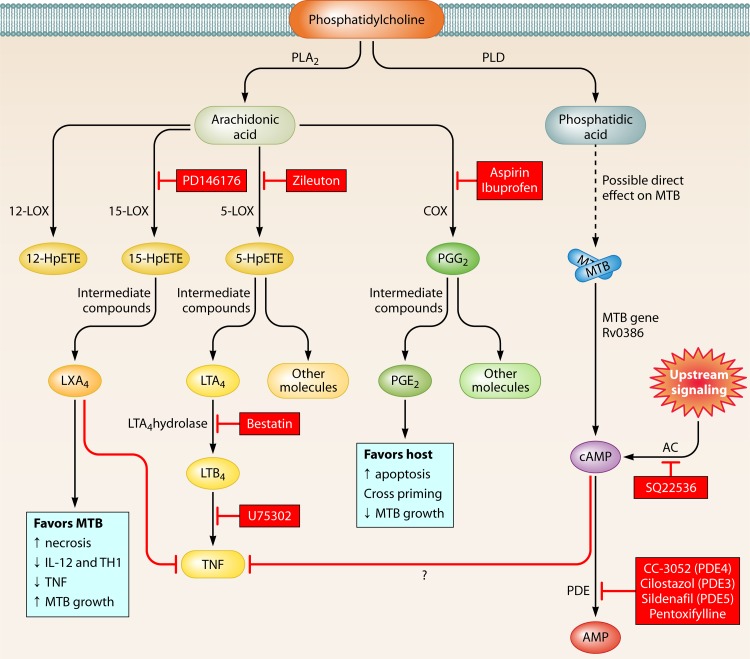
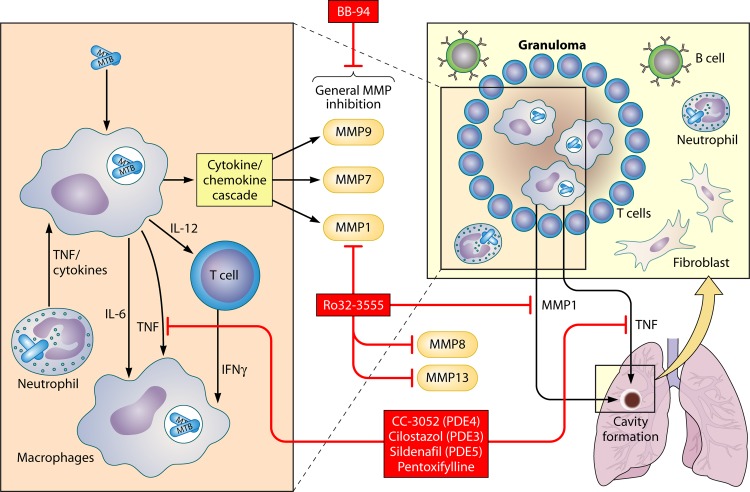
References
-
- Liu PT, Modlin RL. 2008. Human macrophage host defense against Mycobacterium tuberculosis. Curr. Opin. Immunol. 20:371–376 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
