

Primary cilia in neurodevelopmental disorders
- PMID: 24296655
- PMCID: PMC3989897
- DOI: 10.1038/nrneurol.2013.247
Primary cilia in neurodevelopmental disorders
Abstract
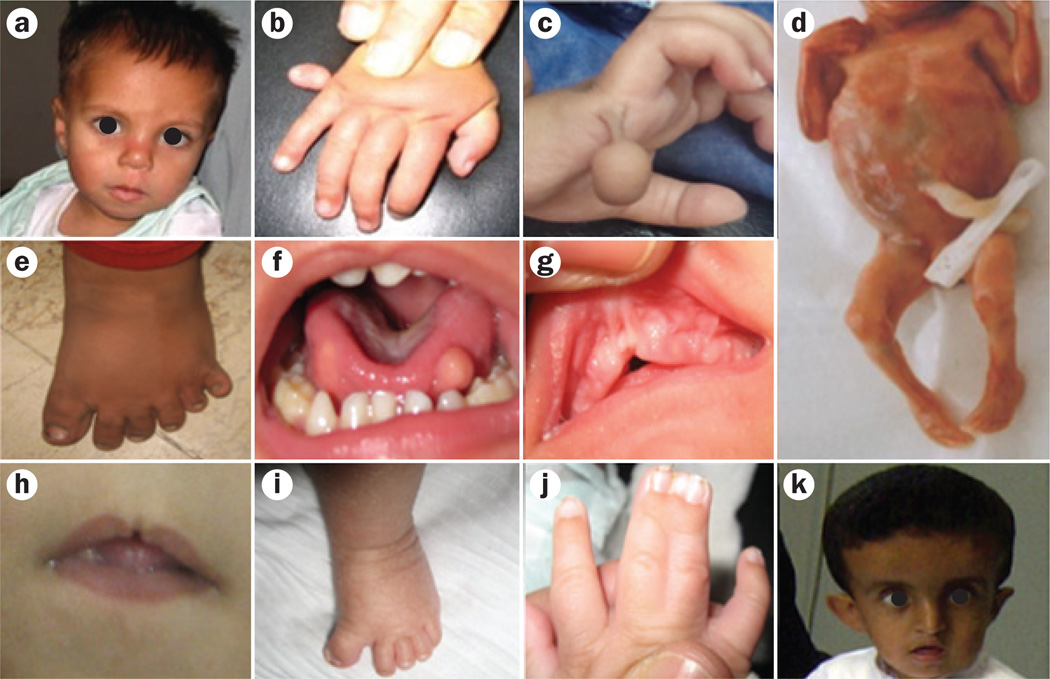
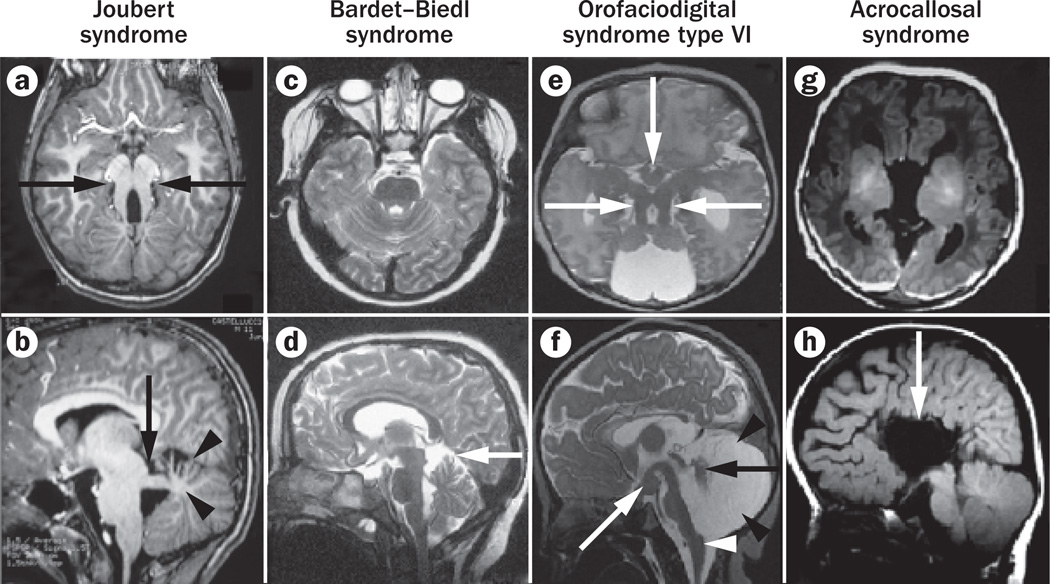
Primary cilia are generally solitary organelles that emanate from the surface of almost all vertebrate cell types. Until recently, details regarding the function of these structures were lacking; however, extensive evidence now suggests that primary cilia have critical roles in sensing the extracellular environment, and in coordinating developmental and homeostatic signalling pathways. Furthermore, disruption of these functions seems to underlie a diverse spectrum of disorders, known as primary ciliopathies. These disorders are characterized by wide-ranging clinical and genetic heterogeneity, but with substantial overlap among distinct conditions. Indeed, ciliopathies are associated with a large variety of manifestations that often include distinctive neurological findings. Herein, we review neurological features associated with primary ciliopathies, highlight genotype-phenotype correlations, and discuss potential mechanisms underlying these findings.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
