

MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4
- PMID: 24297891
- PMCID: PMC3870730
- DOI: 10.1073/pnas.1313944110
MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4
Abstract
Myasthenia gravis (MG) is a severely debilitating autoimmune disease that is due to a decrease in the efficiency of synaptic transmission at neuromuscular synapses. MG is caused by antibodies against postsynaptic proteins, including (i) acetylcholine receptors, the neurotransmitter receptor, (ii) muscle-specific kinase (MuSK), a receptor tyrosine kinase essential for the formation and maintenance of neuromuscular synapses, and (iii) low-density lipoprotein receptor-related protein 4 (Lrp4), which responds to neural Agrin by binding and stimulating MuSK. Passive transfer studies in mice have shown that IgG4 antibodies from MuSK MG patients cause disease without requiring complement or other immune components, suggesting that these MuSK antibodies cause disease by directly interfering with MuSK function. Here we show that pathogenic IgG4 antibodies to MuSK bind to a structural epitope in the first Ig-like domain of MuSK, prevent binding between MuSK and Lrp4, and inhibit Agrin-stimulated MuSK phosphorylation. In contrast, these IgG4 antibodies have no direct effect on MuSK dimerization or MuSK internalization. These results provide insight into the unique pathogenesis of MuSK MG and provide clues toward development of specific treatment options.
Keywords: Dok7; Rapsyn; activation loop; insulin receptor; neuromuscular junction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
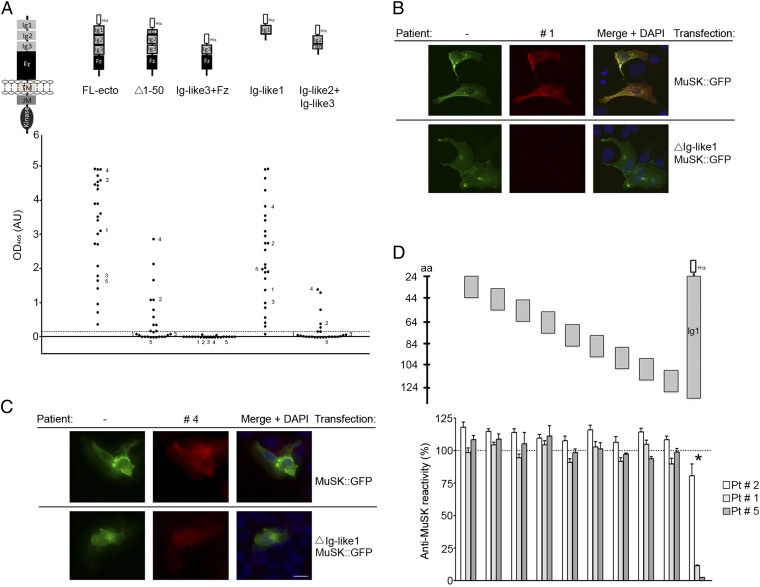
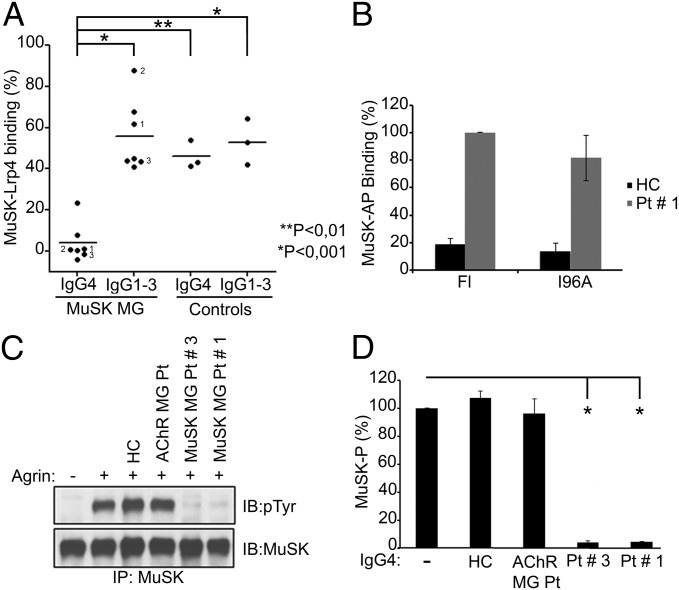
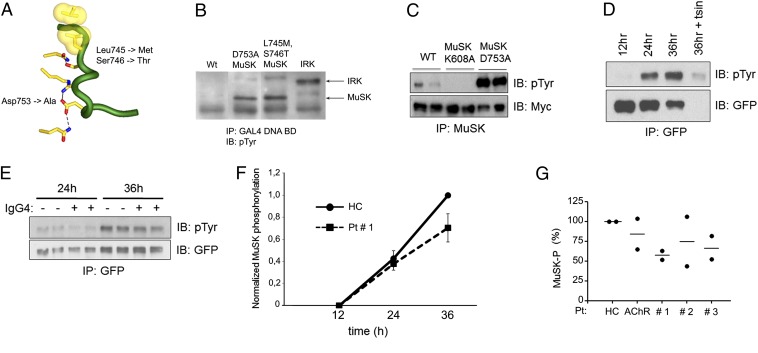
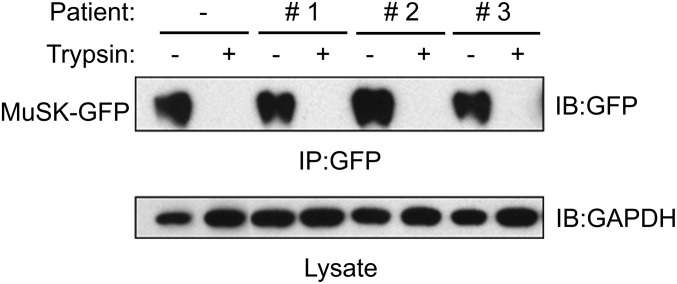
References
-
- Engel AG, Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci. 1987;505:326–332. - PubMed
-
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298(20):1116–1122. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
