

HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase
- PMID: 24299452
- PMCID: PMC4222069
- DOI: 10.1186/1750-1172-8-188
HIBCH mutations can cause Leigh-like disease with combined deficiency of multiple mitochondrial respiratory chain enzymes and pyruvate dehydrogenase
Abstract
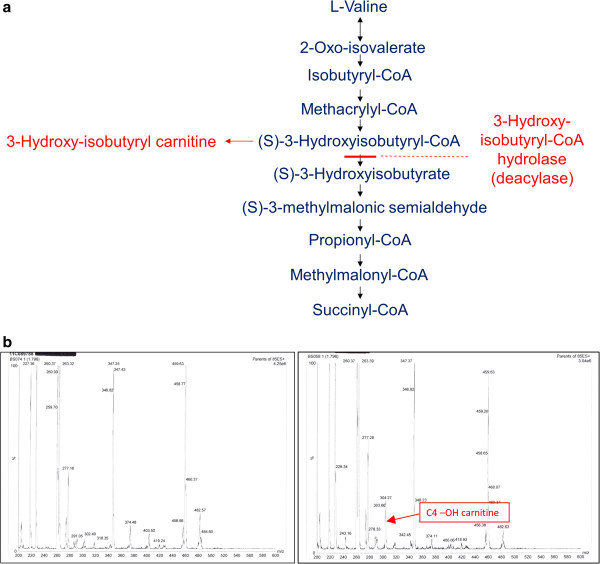
Background: Deficiency of 3-hydroxy-isobutyryl-CoA hydrolase (HIBCH) caused by HIBCH mutations is a rare cerebral organic aciduria caused by disturbance of valine catabolism. Multiple mitochondrial respiratory chain (RC) enzyme deficiencies can arise from a number of mechanisms, including defective maintenance or expression of mitochondrial DNA. Impaired biosynthesis of iron-sulphur clusters and lipoic acid can lead to pyruvate dehydrogenase complex (PDHc) deficiency in addition to multiple RC deficiencies, known as the multiple mitochondrial dysfunctions syndrome.
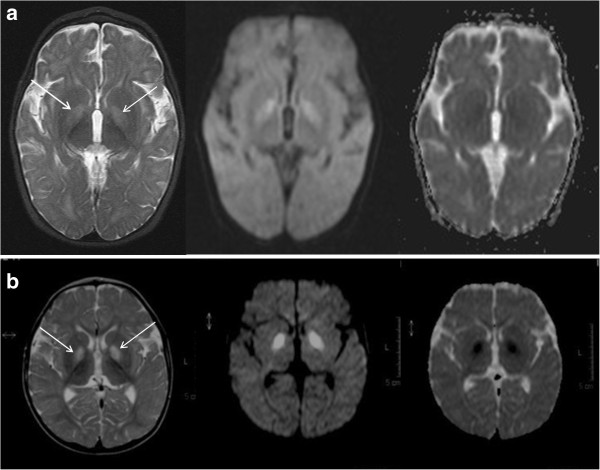
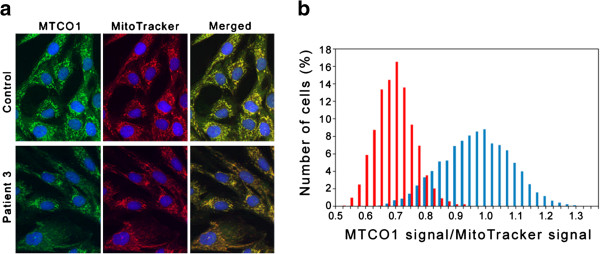
Methods: Two brothers born to distantly related Pakistani parents presenting in early infancy with a progressive neurodegenerative disorder, associated with basal ganglia changes on brain magnetic resonance imaging, were investigated for suspected Leigh-like mitochondrial disease. The index case had deficiencies of multiple RC enzymes and PDHc in skeletal muscle and fibroblasts respectively, but these were normal in his younger brother. The observation of persistently elevated hydroxy-C4-carnitine levels in the younger brother led to suspicion of HIBCH deficiency, which was investigated by biochemical assay in cultured skin fibroblasts and molecular genetic analysis.
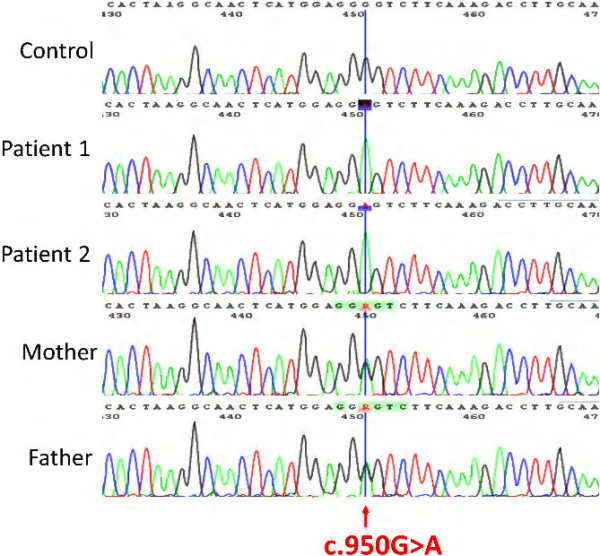
Results: Specific spectrophotometric enzyme assay revealed HIBCH activity to be below detectable limits in cultured skin fibroblasts from both brothers. Direct Sanger sequence analysis demonstrated a novel homozygous pathogenic missense mutation c.950G <A; p.Gly317Glu in the HIBCH gene, which segregated with infantile-onset neurodegeneration within the family.
Conclusions: HIBCH deficiency, a disorder of valine catabolism, is a novel cause of the multiple mitochondrial dysfunctions syndrome, and should be considered in the differential diagnosis of patients presenting with multiple RC deficiencies and/or pyruvate dehydrogenase deficiency.
Figures
References
-
- Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, Del TM, Labayru MT, Landa J, Font A, Garcia-Villoria J. et al.A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–667. doi: 10.1016/j.ajhg.2011.10.005. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
