

From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing
- PMID: 24299736
- PMCID: PMC3941114
- DOI: 10.1101/gr.161034.113
From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing
Abstract
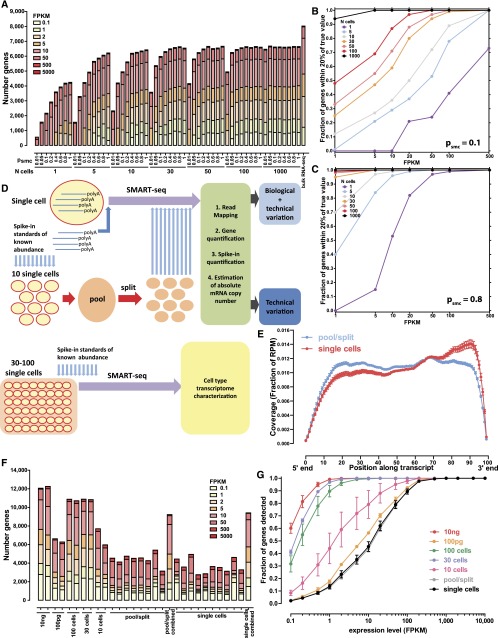
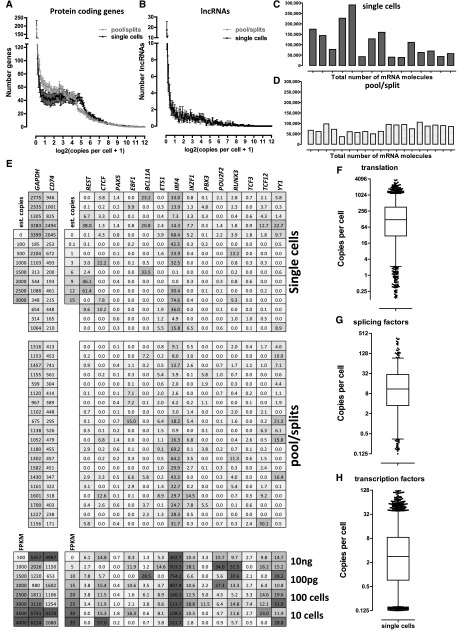
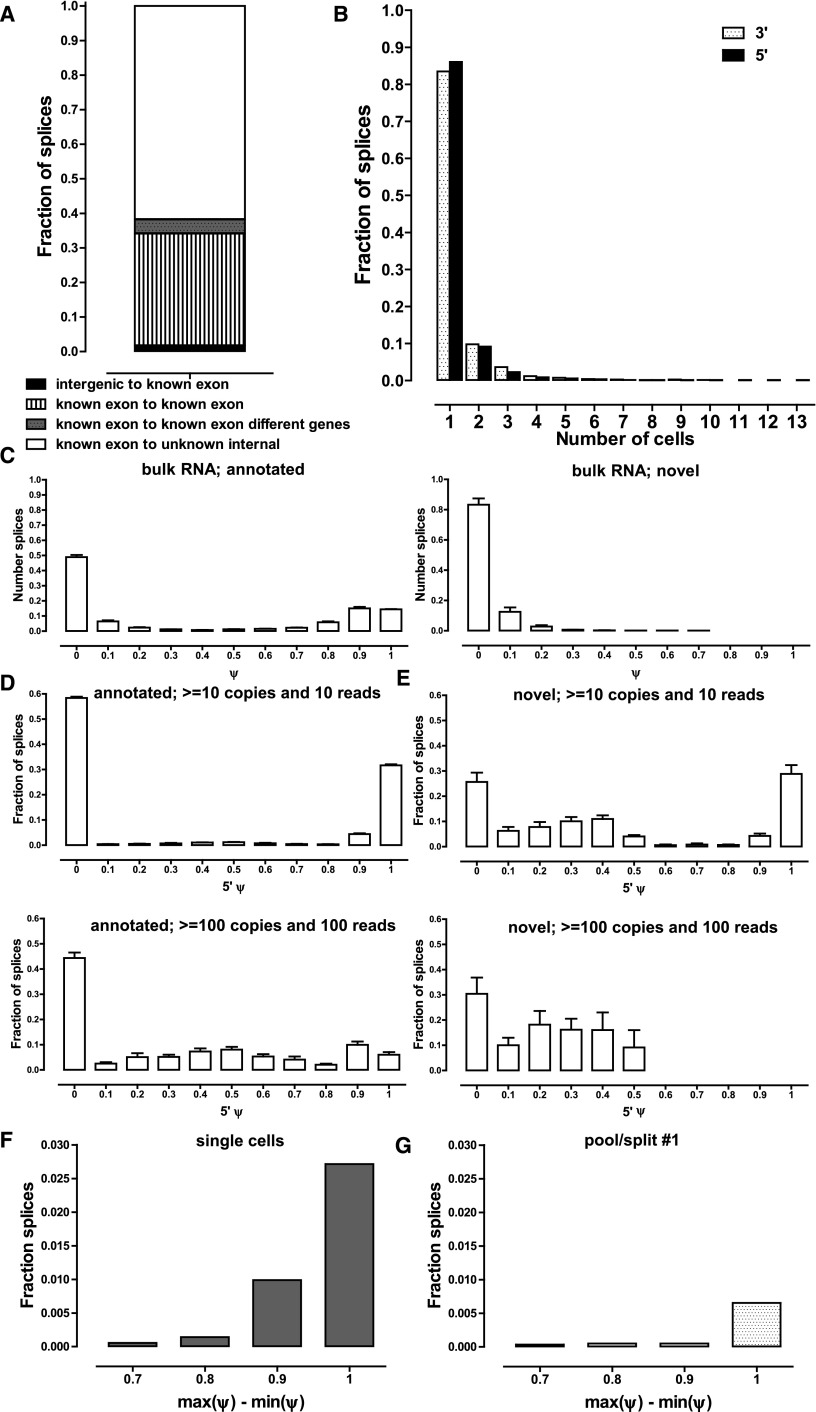
Single-cell RNA-seq mammalian transcriptome studies are at an early stage in uncovering cell-to-cell variation in gene expression, transcript processing and editing, and regulatory module activity. Despite great progress recently, substantial challenges remain, including discriminating biological variation from technical noise. Here we apply the SMART-seq single-cell RNA-seq protocol to study the reference lymphoblastoid cell line GM12878. By using spike-in quantification standards, we estimate the absolute number of RNA molecules per cell for each gene and find significant variation in total mRNA content: between 50,000 and 300,000 transcripts per cell. We directly measure technical stochasticity by a pool/split design and find that there are significant differences in expression between individual cells, over and above technical variation. Specific gene coexpression modules were preferentially expressed in subsets of individual cells, including one enriched for mRNA processing and splicing factors. We assess cell-to-cell variation in alternative splicing and allelic bias and report evidence of significant differences in splice site usage that exceed splice variation in the pool/split comparison. Finally, we show that transcriptomes from small pools of 30-100 cells approach the information content and reproducibility of contemporary RNA-seq from large amounts of input material. Together, our results define an experimental and computational path forward for analyzing gene expression in rare cell types and cell states.
Figures


References
-
- Blake WJ, Kaern M, Cantor CR, Collins JJ 2003. Noise in eukaryotic gene expression. Nature 422: 633–637 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases