

Comparison of software packages for detecting differential expression in RNA-seq studies
- PMID: 24300110
- PMCID: PMC4293378
- DOI: 10.1093/bib/bbt086
Comparison of software packages for detecting differential expression in RNA-seq studies
Abstract
RNA-sequencing (RNA-seq) has rapidly become a popular tool to characterize transcriptomes. A fundamental research problem in many RNA-seq studies is the identification of reliable molecular markers that show differential expression between distinct sample groups. Together with the growing popularity of RNA-seq, a number of data analysis methods and pipelines have already been developed for this task. Currently, however, there is no clear consensus about the best practices yet, which makes the choice of an appropriate method a daunting task especially for a basic user without a strong statistical or computational background. To assist the choice, we perform here a systematic comparison of eight widely used software packages and pipelines for detecting differential expression between sample groups in a practical research setting and provide general guidelines for choosing a robust pipeline. In general, our results demonstrate how the data analysis tool utilized can markedly affect the outcome of the data analysis, highlighting the importance of this choice.
Keywords: RNA-seq; differential expression; gene expression.
© The Author 2013. Published by Oxford University Press.
Figures




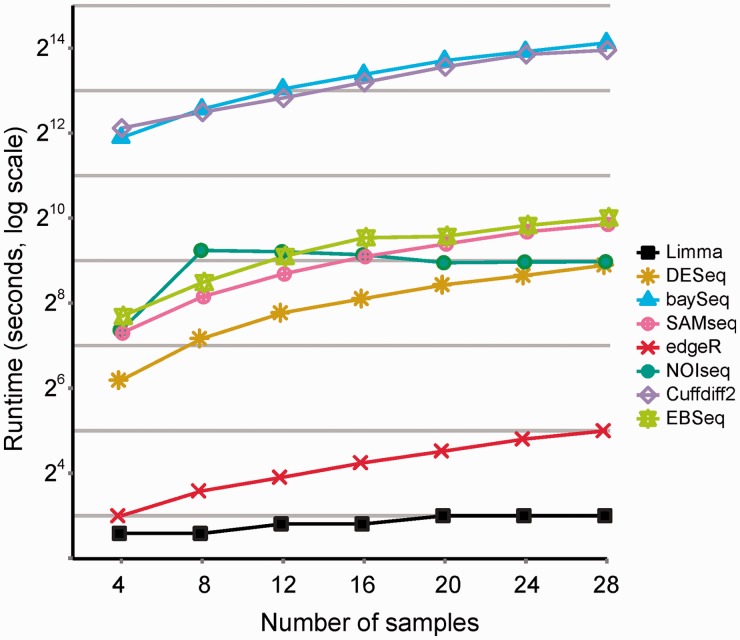
References
-
- Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8:469–77. - PubMed
-
- Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
