

Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids
- PMID: 24302004
- PMCID: PMC3920006
- DOI: 10.1152/ajpendo.00202.2013
Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids
Erratum in
-
Corrigendum.Am J Physiol Endocrinol Metab. 2016 Nov 1;311(5):E899. doi: 10.1152/ajpendo.zh1-7676-corr.2016. Am J Physiol Endocrinol Metab. 2016. PMID: 27888188 Free PMC article. No abstract available.
Retraction in
-
Retraction for Li et al., volume 306, 2014, p. E197-E209.Am J Physiol Endocrinol Metab. 2022 Oct 1;323(4):E403. doi: 10.1152/ajpendo.00202.2013_ret. Am J Physiol Endocrinol Metab. 2022. PMID: 36219585 Free PMC article. No abstract available.
Abstract
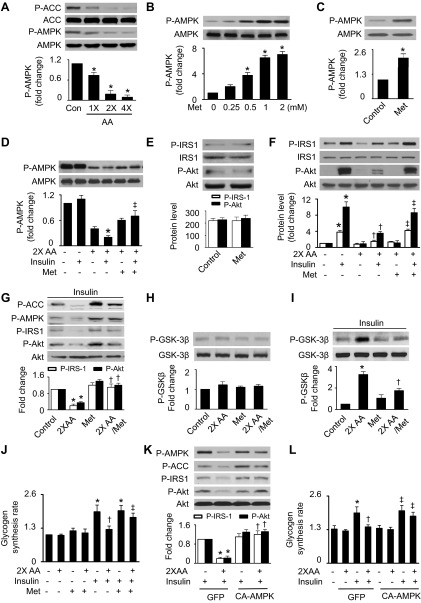
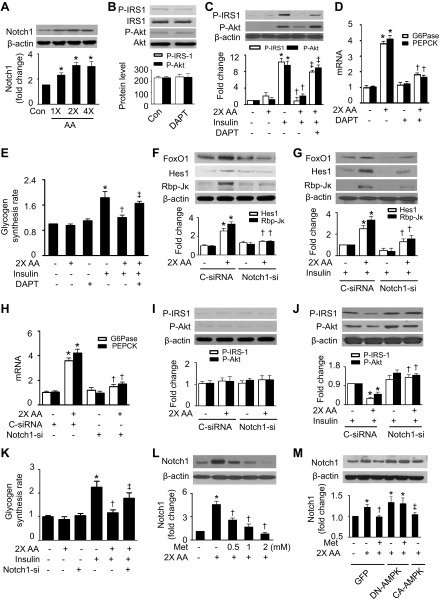
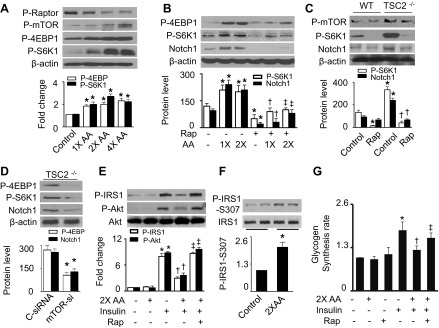
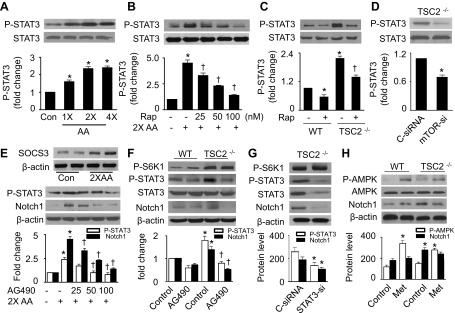
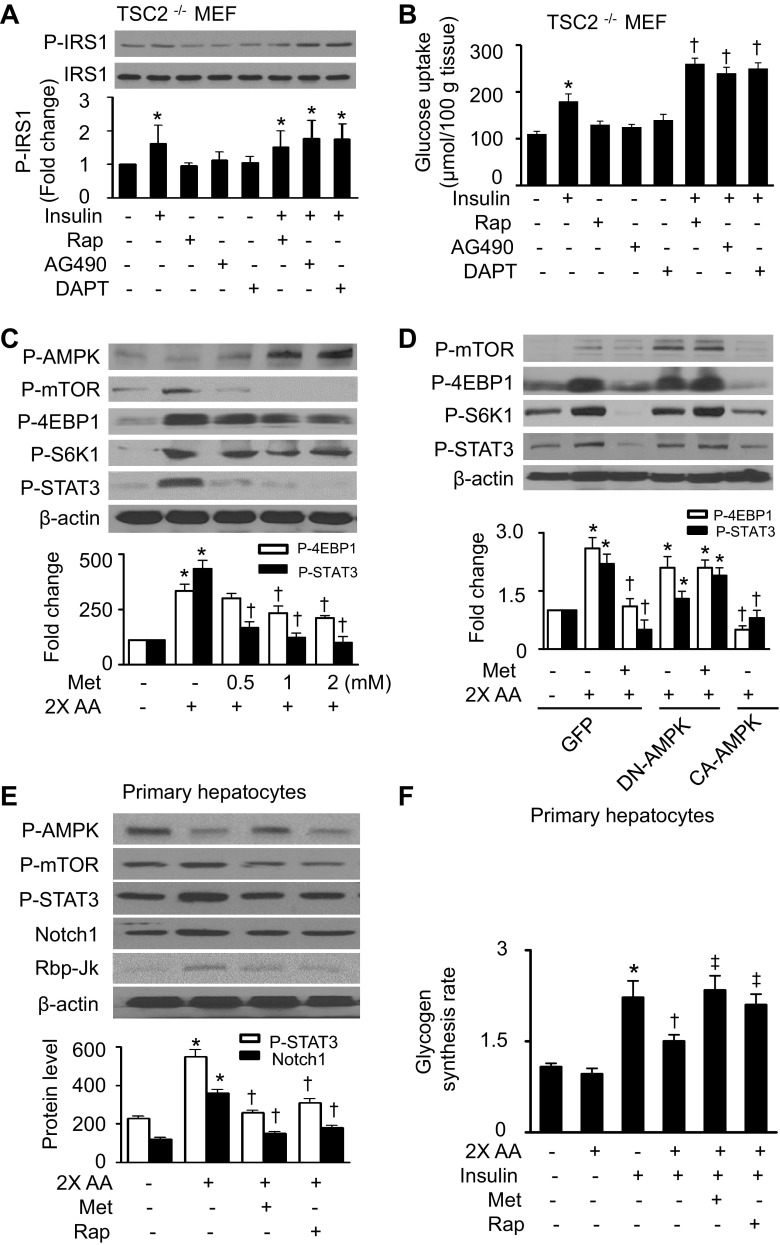
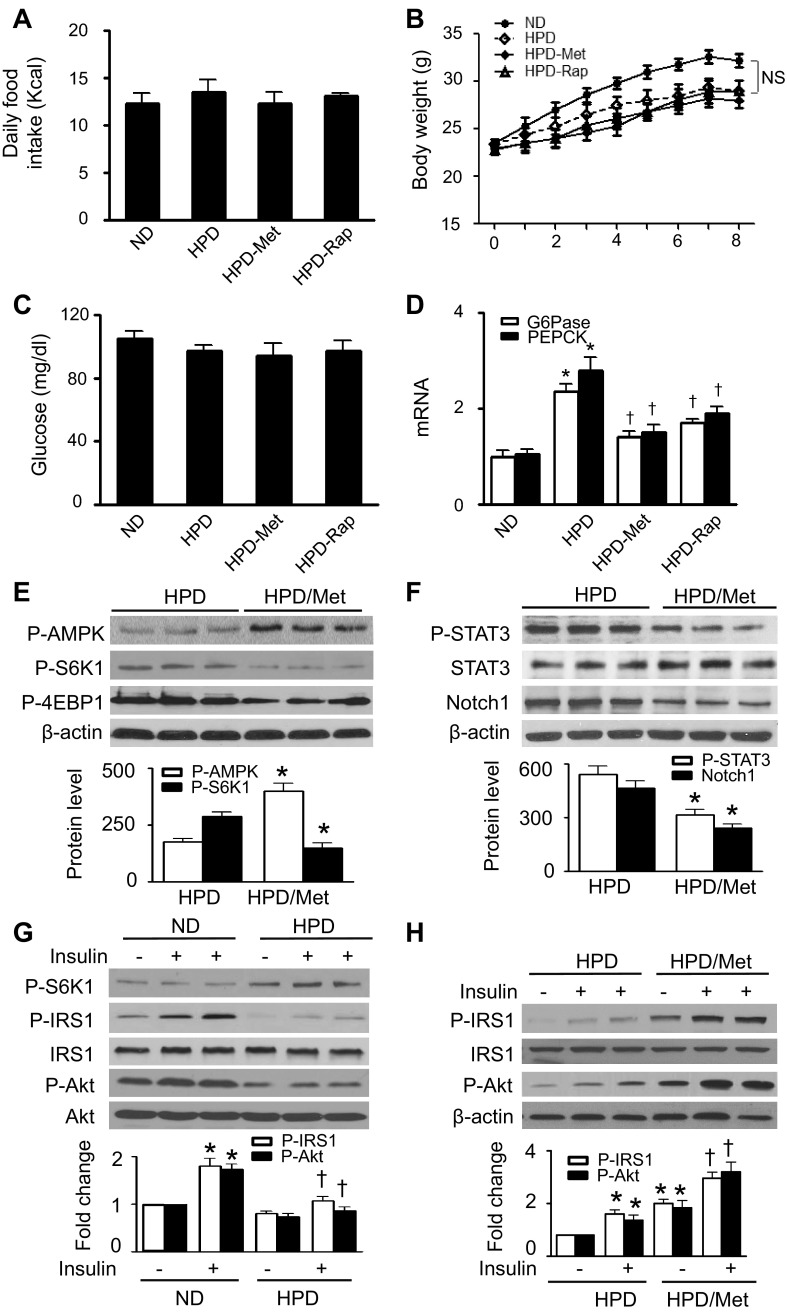
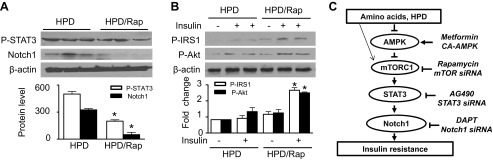
Nutrient overload is associated with the development of obesity, insulin resistance, and type 2 diabetes. However, the underlying mechanisms for developing insulin resistance in the presence of excess nutrients are incompletely understood. We investigated whether activation of AMP-activated protein kinase (AMPK) prevents the hepatic insulin resistance that is induced by the consumption of a high-protein diet (HPD) and the presence of excess amino acids. Exposure of HepG2 cells to excess amino acids reduced AMPK phosphorylation, upregulated Notch1 expression, and impaired the insulin-stimulated phosphorylation of Akt Ser(473) and insulin receptor substrate-1 (IRS-1) Tyr(612). Inhibition of Notch1 prevented amino acid-induced insulin resistance, which was accompanied by reduced expression of Rbp-Jk, hairy and enhancer of split-1, and forkhead box O1. Mechanistically, mTORC1 signaling was activated by excess amino acids, which then positively regulated Notch1 expression through the activation of the signal transducer and activator of transcription 3 (STAT3). Activation of AMPK by metformin inhibited mTORC1-STAT3 signaling, thereby preventing excess amino acid-impaired insulin signaling. Finally, HPD feeding suppressed AMPK activity, activated mTORC1/STAT3/Notch1 signaling, and induced insulin resistance. Chronic administration of either metformin or rapamycin inhibited the HPD-activated mTORC1/STAT3/Notch1 signaling pathway and prevented hepatic insulin resistance. We conclude that the upregulation of Notch1 expression by hyperactive mTORC1 signaling is an essential event in the development of hepatic insulin resistance in the presence of excess amino acids. Activation of AMPK prevents amino acid-induced insulin resistance through the suppression of the mTORC1/STAT3/Notch1 signaling pathway.
Keywords: AMP-activated protein kinase; Notch; amino acids; insulin resistance; mammalian target of rapamycin complex 1; signal transducer and activator of transcription 3.
Figures
Similar articles
-
AMPK activation prevents excess nutrient-induced hepatic lipid accumulation by inhibiting mTORC1 signaling and endoplasmic reticulum stress response.Biochim Biophys Acta. 2014 Sep;1842(9):1844-54. doi: 10.1016/j.bbadis.2014.07.002. Epub 2014 Jul 10. Biochim Biophys Acta. 2014. PMID: 25016145 Free PMC article.
-
Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation.J Biol Chem. 2009 Dec 18;284(51):35425-32. doi: 10.1074/jbc.M109.051516. J Biol Chem. 2009. PMID: 19875458 Free PMC article.
-
The peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells.Diabetologia. 2012 Mar;55(3):743-51. doi: 10.1007/s00125-011-2401-4. Epub 2011 Dec 17. Diabetologia. 2012. PMID: 22179221
-
New insights into Notch1 regulation of the PI3K-AKT-mTOR1 signaling axis: targeted therapy of γ-secretase inhibitor resistant T-cell acute lymphoblastic leukemia.Cell Signal. 2014 Jan;26(1):149-61. doi: 10.1016/j.cellsig.2013.09.021. Epub 2013 Oct 16. Cell Signal. 2014. PMID: 24140475 Review.
-
The lysosome: a crucial hub for AMPK and mTORC1 signalling.Biochem J. 2017 Apr 13;474(9):1453-1466. doi: 10.1042/BCJ20160780. Biochem J. 2017. PMID: 28408430 Review.
Cited by
-
Triclosan induces ROS-dependent cell death and autophagy in A375 melanoma cells.Oncol Lett. 2020 Oct;20(4):73. doi: 10.3892/ol.2020.11934. Epub 2020 Jul 30. Oncol Lett. 2020. PMID: 32863906 Free PMC article.
-
Metformin Reduces Desmoplasia in Pancreatic Cancer by Reprogramming Stellate Cells and Tumor-Associated Macrophages.PLoS One. 2015 Dec 7;10(12):e0141392. doi: 10.1371/journal.pone.0141392. eCollection 2015. PLoS One. 2015. PMID: 26641266 Free PMC article.
-
Autophagy-induced degradation of Notch1, achieved through intermittent fasting, may promote beta cell neogenesis: implications for reversal of type 2 diabetes.Open Heart. 2019 May 22;6(1):e001028. doi: 10.1136/openhrt-2019-001028. eCollection 2019. Open Heart. 2019. PMID: 31218007 Free PMC article. No abstract available.
-
AMPK activation prevents excess nutrient-induced hepatic lipid accumulation by inhibiting mTORC1 signaling and endoplasmic reticulum stress response.Biochim Biophys Acta. 2014 Sep;1842(9):1844-54. doi: 10.1016/j.bbadis.2014.07.002. Epub 2014 Jul 10. Biochim Biophys Acta. 2014. PMID: 25016145 Free PMC article.
-
Effect of a high dose atorvastatin as added-on therapy on symptoms and serum AMPK/NLRP3 inflammasome and IL-6/STAT3 axes in patients with major depressive disorder: randomized controlled clinical study.Front Pharmacol. 2024 May 24;15:1381523. doi: 10.3389/fphar.2024.1381523. eCollection 2024. Front Pharmacol. 2024. PMID: 38855751 Free PMC article.
References
-
- Adibi SA. Influence of dietary deprivations on plasma concentration of free amino acids of man. J Appl Physiol 25: 52–57, 1968 - PubMed
-
- Armstrong JL, Bonavaud SM, Toole BJ, Yeaman SJ. Regulation of glycogen synthesis by amino acids in cultured human muscle cells. J Biol Chem 276: 952–956, 2001 - PubMed
-
- Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol 197: 157–168, 2003 - PubMed
-
- Carling D. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci 29: 18–24, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL089920/HL/NHLBI NIH HHS/United States
- R01 HL074399/HL/NHLBI NIH HHS/United States
- R01 AG047776/AG/NIA NIH HHS/United States
- 1P20-RR-024215-01/RR/NCRR NIH HHS/United States
- R01 HL079584/HL/NHLBI NIH HHS/United States
- HL-080499/HL/NHLBI NIH HHS/United States
- HL-074399/HL/NHLBI NIH HHS/United States
- R01 HL080499/HL/NHLBI NIH HHS/United States
- HL-096032/HL/NHLBI NIH HHS/United States
- HL-079584/HL/NHLBI NIH HHS/United States
- R01 HL110488/HL/NHLBI NIH HHS/United States
- R01 HL105157/HL/NHLBI NIH HHS/United States
- HL-089920/HL/NHLBI NIH HHS/United States
- R01 HL096032/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous