

The complex methylome of the human gastric pathogen Helicobacter pylori
- PMID: 24302578
- PMCID: PMC3936762
- DOI: 10.1093/nar/gkt1201
The complex methylome of the human gastric pathogen Helicobacter pylori
Abstract
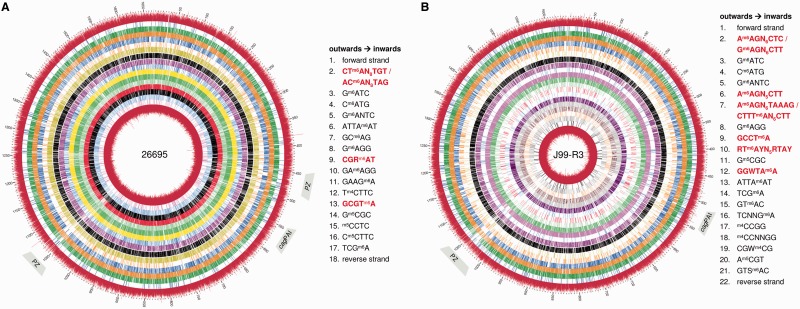
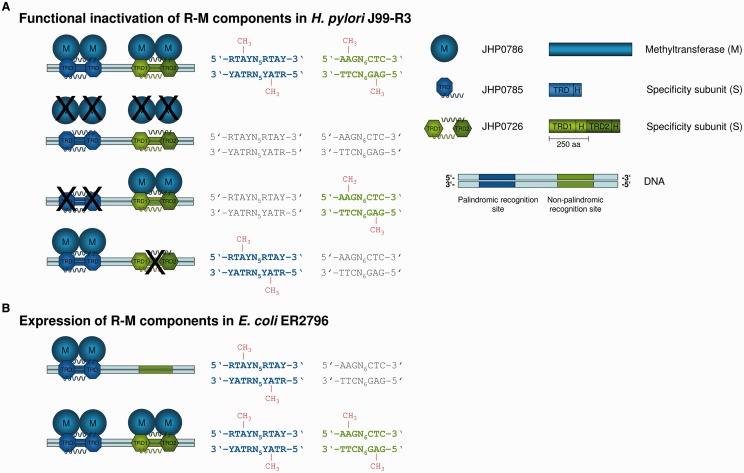
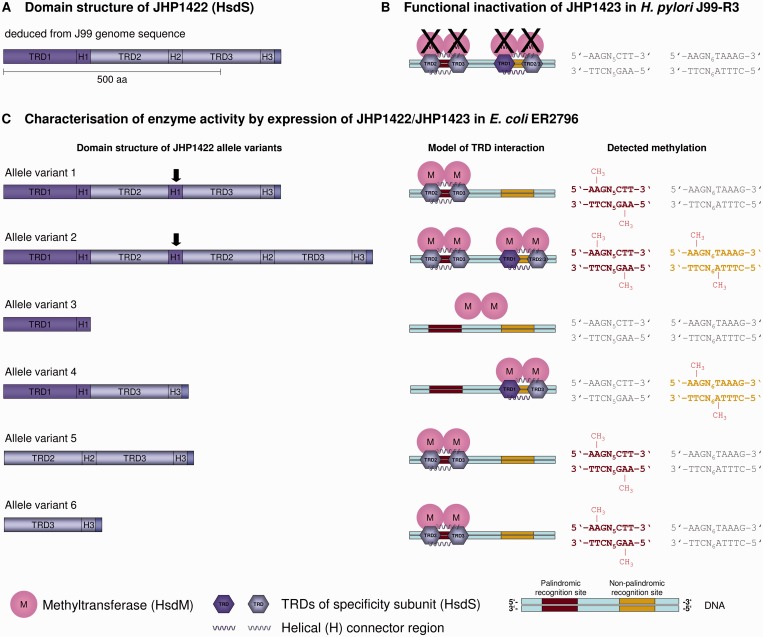
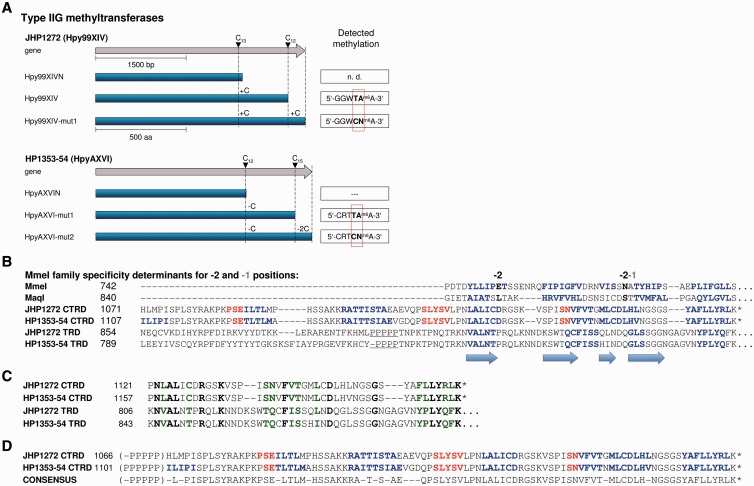
The genome of Helicobacter pylori is remarkable for its large number of restriction-modification (R-M) systems, and strain-specific diversity in R-M systems has been suggested to limit natural transformation, the major driving force of genetic diversification in H. pylori. We have determined the comprehensive methylomes of two H. pylori strains at single base resolution, using Single Molecule Real-Time (SMRT®) sequencing. For strains 26695 and J99-R3, 17 and 22 methylated sequence motifs were identified, respectively. For most motifs, almost all sites occurring in the genome were detected as methylated. Twelve novel methylation patterns corresponding to nine recognition sequences were detected (26695, 3; J99-R3, 6). Functional inactivation, correction of frameshifts as well as cloning and expression of candidate methyltransferases (MTases) permitted not only the functional characterization of multiple, yet undescribed, MTases, but also revealed novel features of both Type I and Type II R-M systems, including frameshift-mediated changes of sequence specificity and the interaction of one MTase with two alternative specificity subunits resulting in different methylation patterns. The methylomes of these well-characterized H. pylori strains will provide a valuable resource for future studies investigating the role of H. pylori R-M systems in limiting transformation as well as in gene regulation and host interaction.
Figures

Similar articles
-
The Helicobacter pylori Methylome: Roles in Gene Regulation and Virulence.Curr Top Microbiol Immunol. 2017;400:105-127. doi: 10.1007/978-3-319-50520-6_5. Curr Top Microbiol Immunol. 2017. PMID: 28124151 Review.
-
The complete methylome of Helicobacter pylori UM032.BMC Genomics. 2015 Jun 2;16(1):424. doi: 10.1186/s12864-015-1585-2. BMC Genomics. 2015. PMID: 26031894 Free PMC article.
-
Functional analysis of putative restriction-modification system genes in the Helicobacter pylori J99 genome.Nucleic Acids Res. 2000 Sep 1;28(17):3216-23. doi: 10.1093/nar/28.17.3216. Nucleic Acids Res. 2000. PMID: 10954588 Free PMC article.
-
Methylome diversification through changes in DNA methyltransferase sequence specificity.PLoS Genet. 2014 Apr 10;10(4):e1004272. doi: 10.1371/journal.pgen.1004272. eCollection 2014 Apr. PLoS Genet. 2014. PMID: 24722038 Free PMC article.
-
[Diversity in genome and epigenome of Helicobacter pylori].Nihon Saikingaku Zasshi. 2015;70(4):383-9. doi: 10.3412/jsb.70.383. Nihon Saikingaku Zasshi. 2015. PMID: 26632218 Review. Japanese.
Cited by
-
Helicobacter pylori: Genomic Insight into the Host-Pathogen Interaction.Int J Genomics. 2015;2015:386905. doi: 10.1155/2015/386905. Epub 2015 Feb 5. Int J Genomics. 2015. PMID: 25722969 Free PMC article. Review.
-
Novel Methyltransferase Recognition Motif Identified in Chania multitudinisentens RB-25(T) gen. nov., sp. nov.Front Microbiol. 2016 Aug 31;7:1362. doi: 10.3389/fmicb.2016.01362. eCollection 2016. Front Microbiol. 2016. PMID: 27630623 Free PMC article. No abstract available.
-
Kinetic and catalytic properties of M.HpyAXVII, a phase-variable DNA methyltransferase from Helicobacter pylori.J Biol Chem. 2019 Jan 18;294(3):1019-1034. doi: 10.1074/jbc.RA118.003769. Epub 2018 Nov 26. J Biol Chem. 2019. PMID: 30478171 Free PMC article.
-
Clinical microbiology informatics.Clin Microbiol Rev. 2014 Oct;27(4):1025-47. doi: 10.1128/CMR.00049-14. Clin Microbiol Rev. 2014. PMID: 25278581 Free PMC article. Review.
-
DNA methylome regulates virulence and metabolism in Pseudomonas syringae.Elife. 2025 Feb 24;13:RP96290. doi: 10.7554/eLife.96290. Elife. 2025. PMID: 39992965 Free PMC article.
References
-
- Suerbaum S, Michetti P. Helicobacter pylori infection. N. Engl. J. Med. 2002;347:1175–1186. - PubMed
-
- Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. - PubMed
-
- Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007;5:441–452. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
