

Vespucci: a system for building annotated databases of nascent transcripts
- PMID: 24304890
- PMCID: PMC3936758
- DOI: 10.1093/nar/gkt1237
Vespucci: a system for building annotated databases of nascent transcripts
Abstract
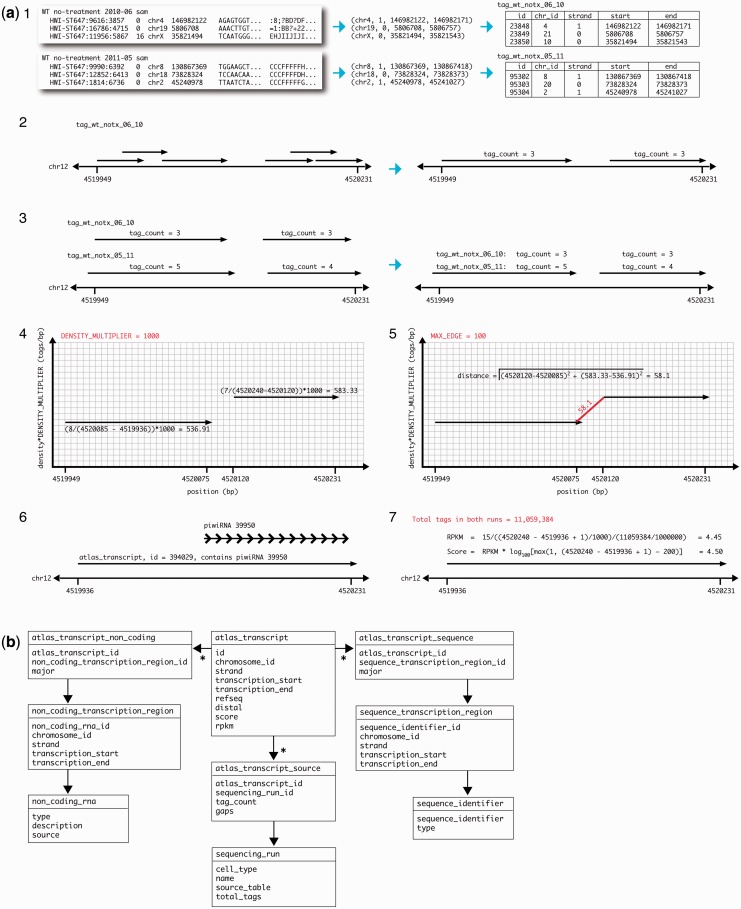
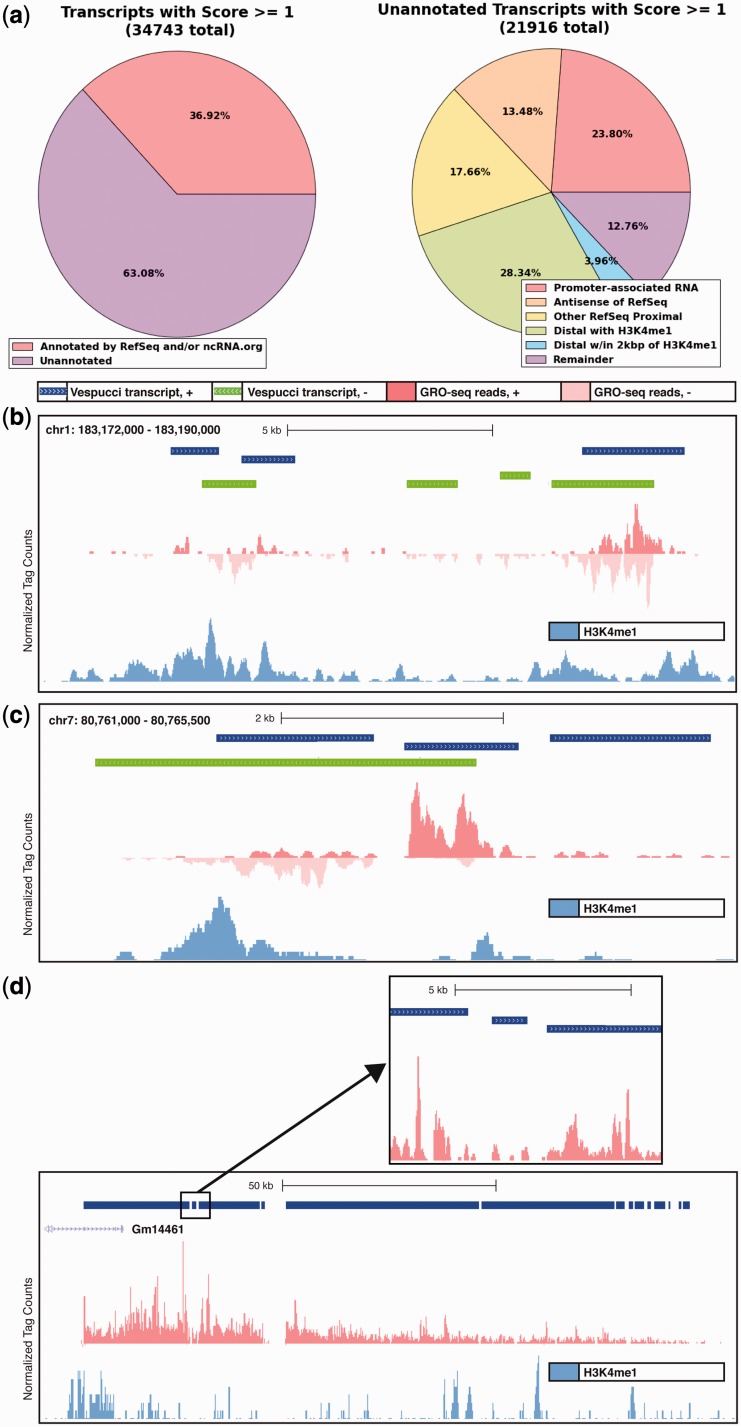
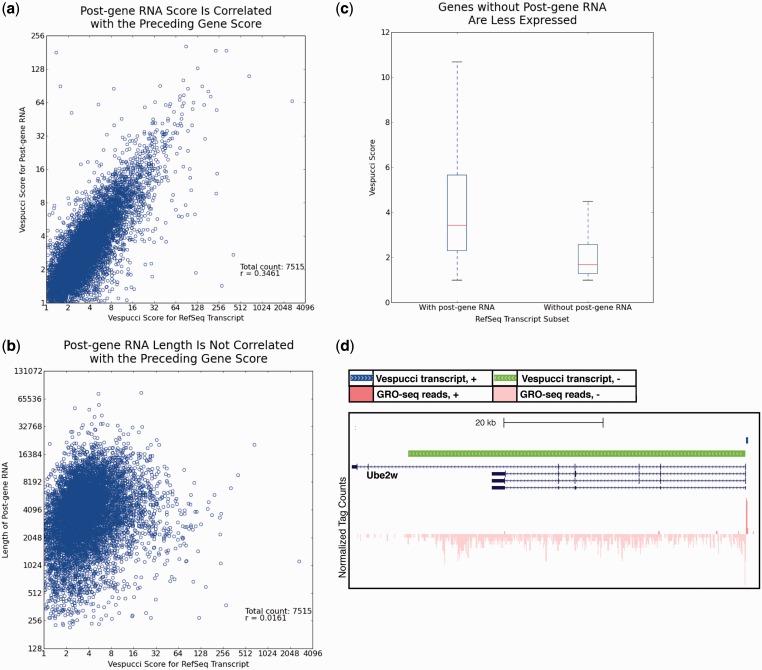
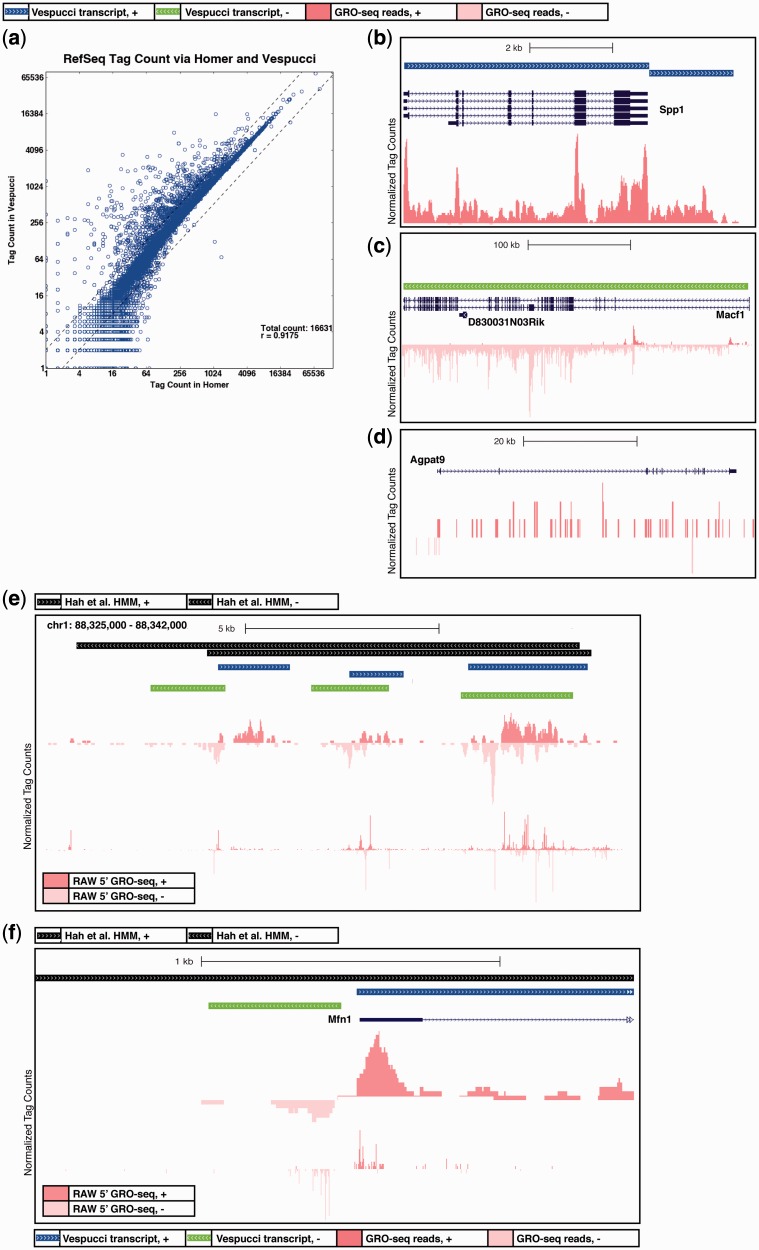
Global run-on sequencing (GRO-seq) is a recent addition to the series of high-throughput sequencing methods that enables new insights into transcriptional dynamics within a cell. However, GRO-sequencing presents new algorithmic challenges, as existing analysis platforms for ChIP-seq and RNA-seq do not address the unique problem of identifying transcriptional units de novo from short reads located all across the genome. Here, we present a novel algorithm for de novo transcript identification from GRO-sequencing data, along with a system that determines transcript regions, stores them in a relational database and associates them with known reference annotations. We use this method to analyze GRO-sequencing data from primary mouse macrophages and derive novel quantitative insights into the extent and characteristics of non-coding transcription in mammalian cells. In doing so, we demonstrate that Vespucci expands existing annotations for mRNAs and lincRNAs by defining the primary transcript beyond the polyadenylation site. In addition, Vespucci generates assemblies for un-annotated non-coding RNAs such as those transcribed from enhancer-like elements. Vespucci thereby provides a robust system for defining, storing and analyzing diverse classes of primary RNA transcripts that are of increasing biological interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
