

Congenital muscular dystrophies and congenital myopathies
- PMID: 24305446
- PMCID: PMC10564049
- DOI: 10.1212/01.CON.0000440658.03557.f1
Congenital muscular dystrophies and congenital myopathies
Abstract
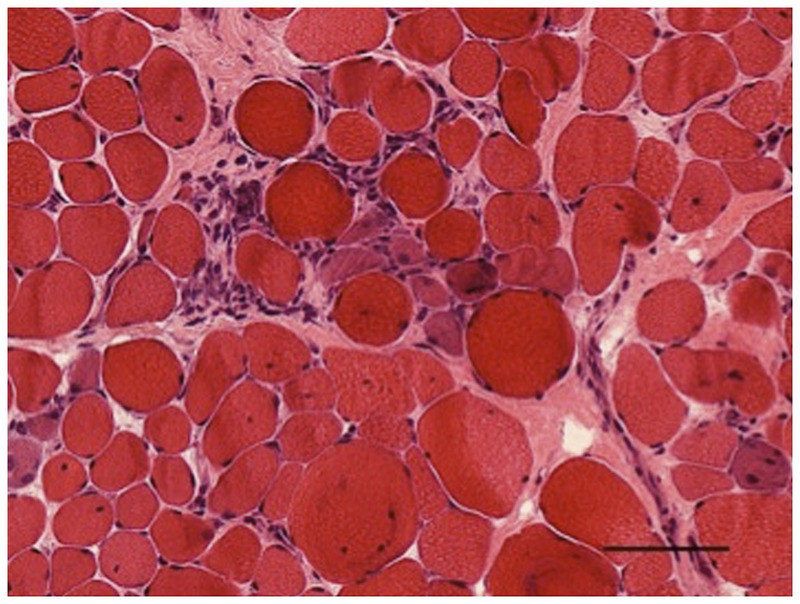
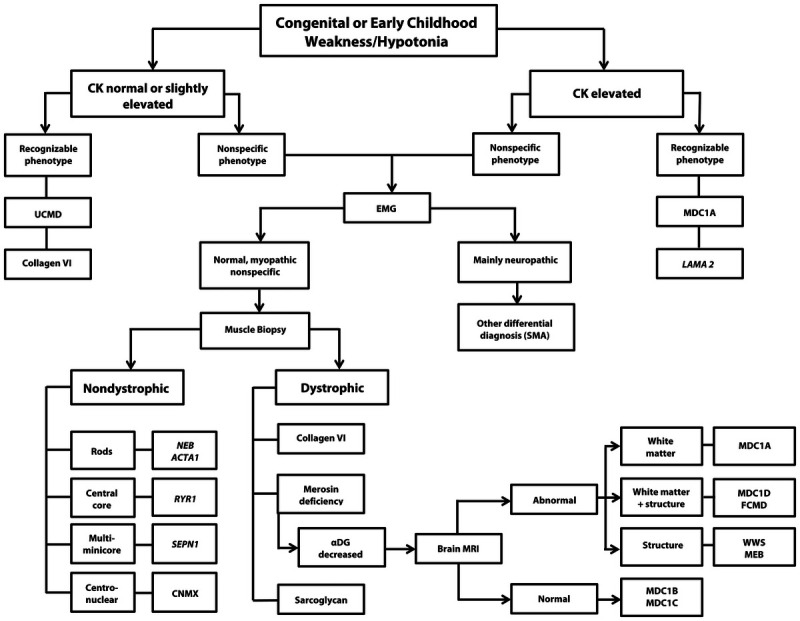
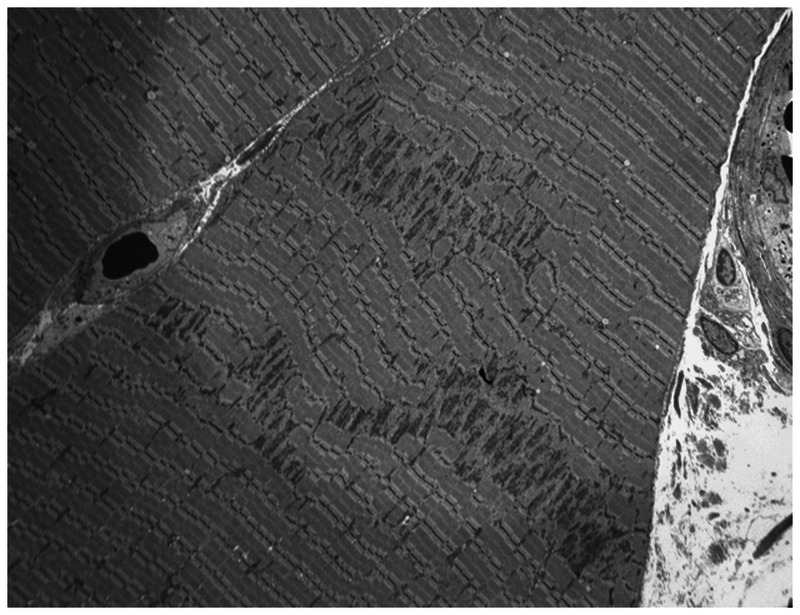
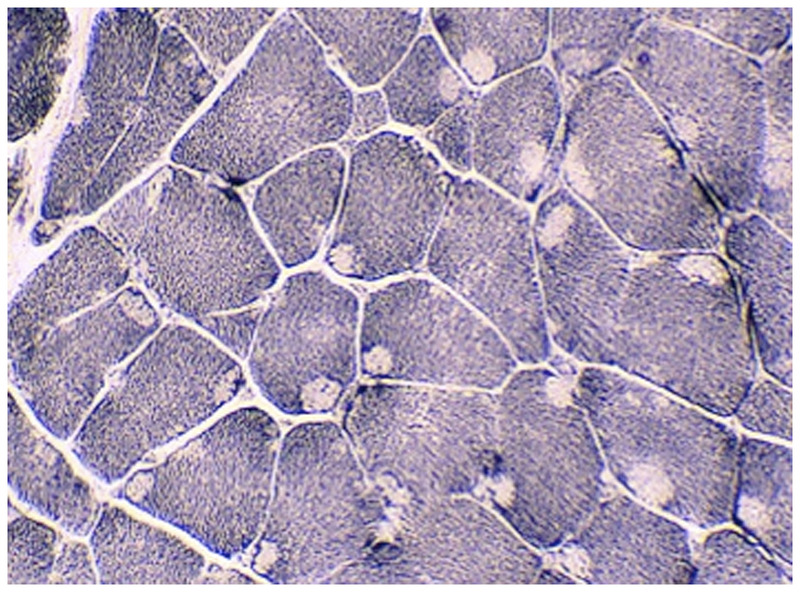
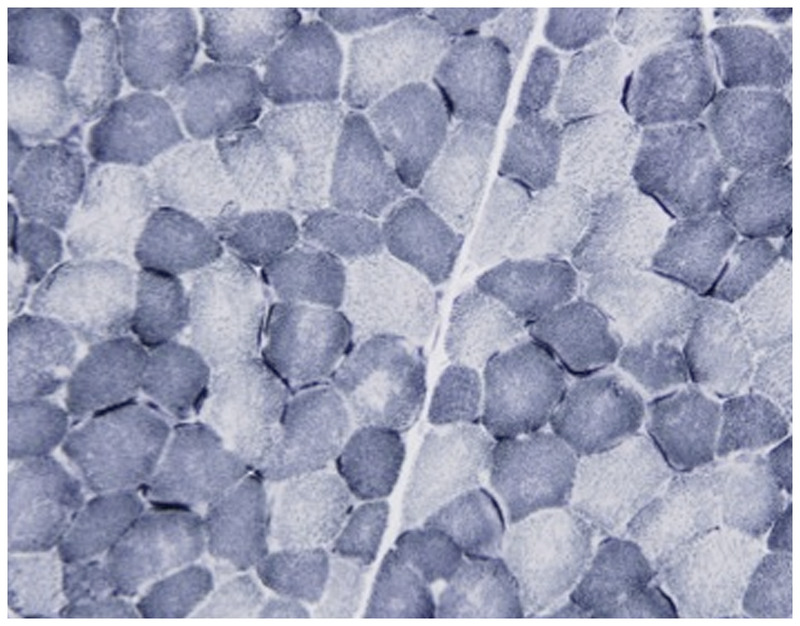
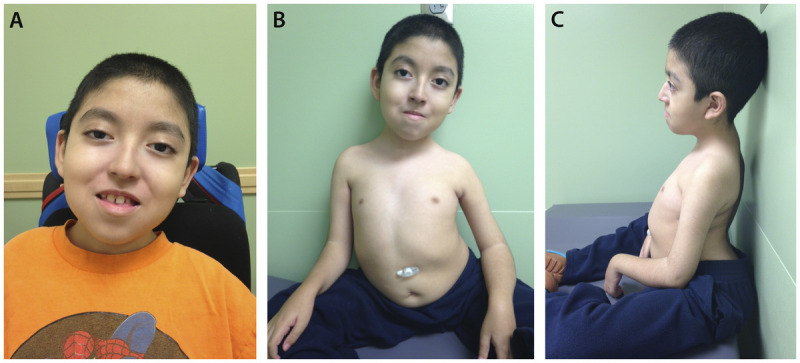
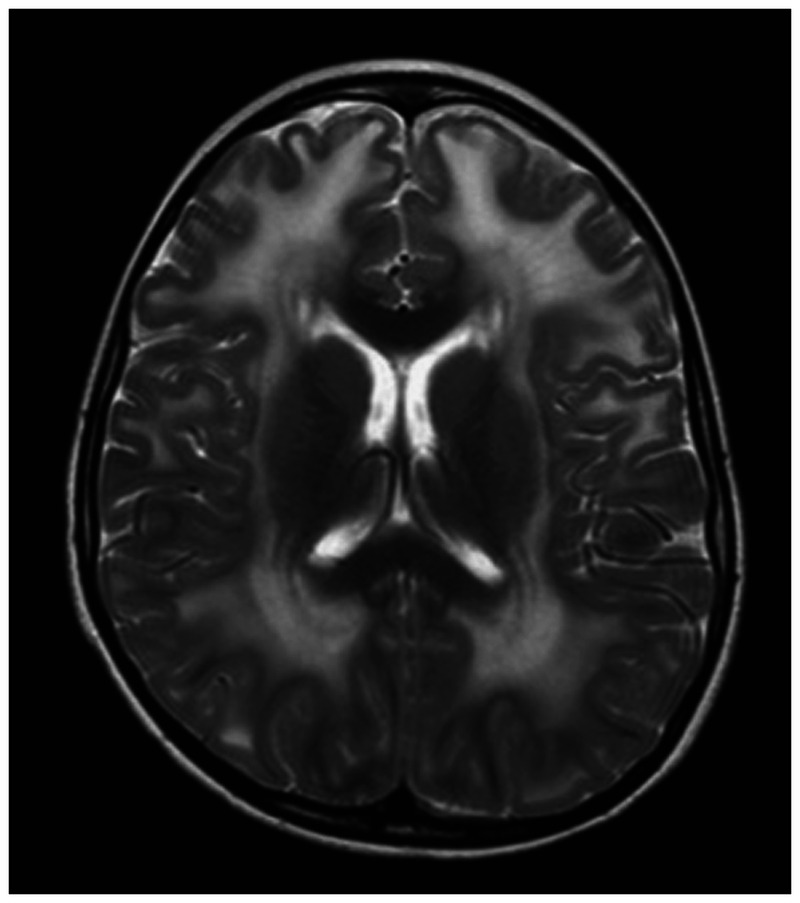
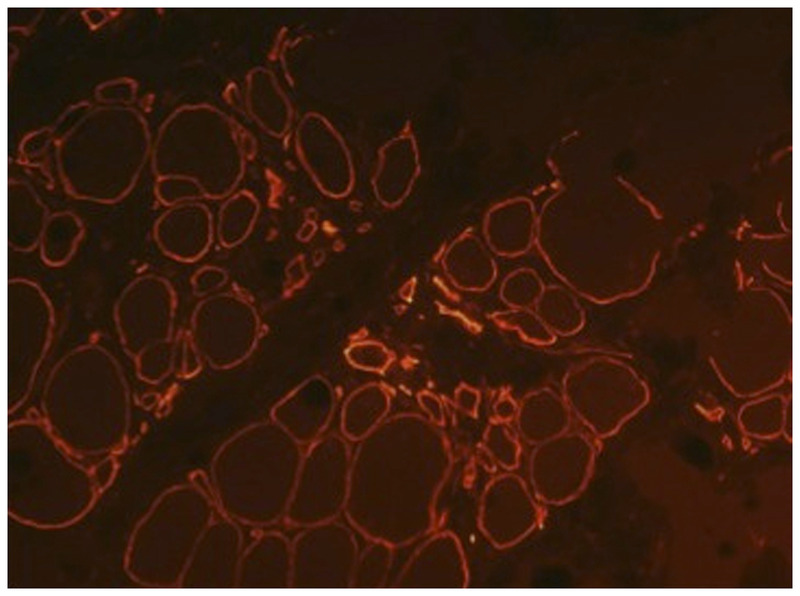
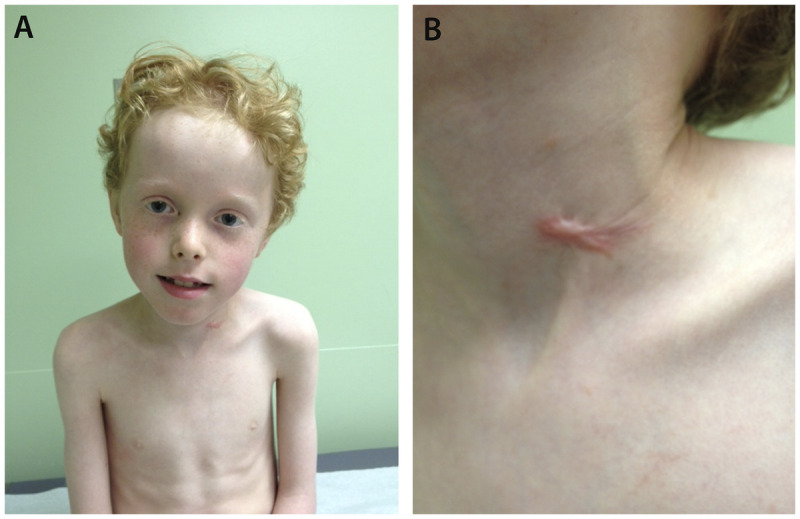
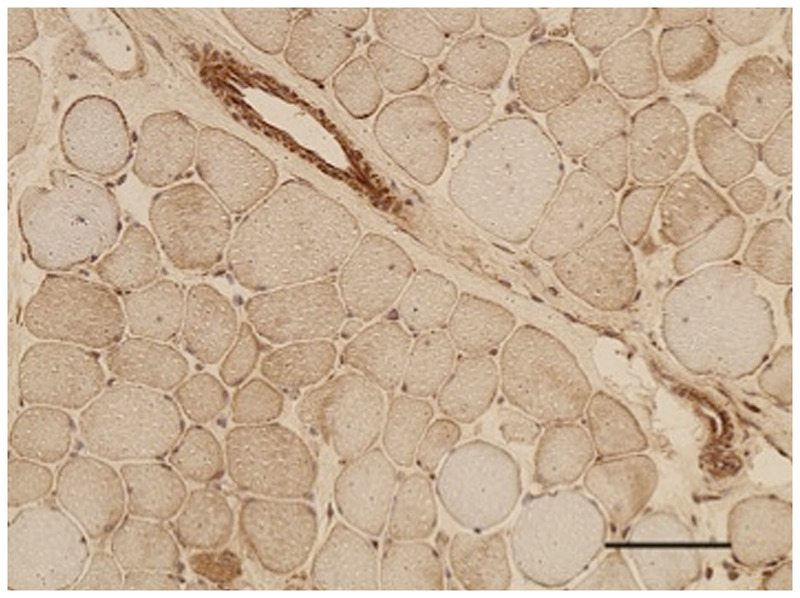
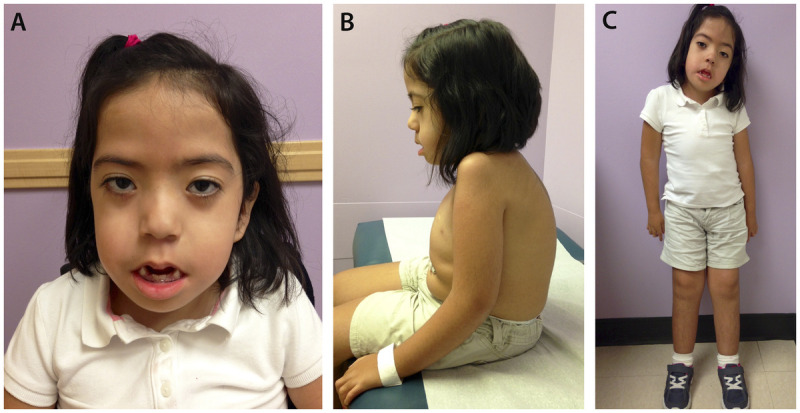
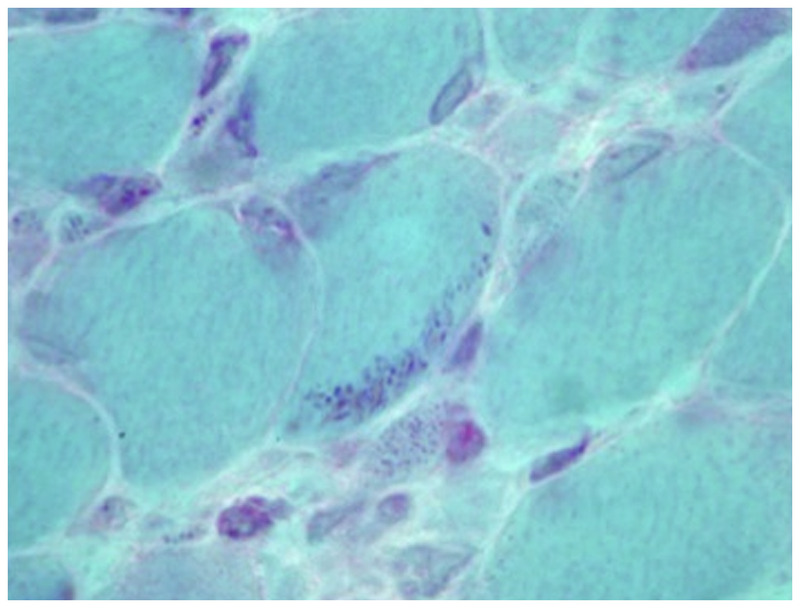
Purpose of review: The purpose of this review is to provide information regarding the diagnosis and natural history of some very rare disorders: congenital muscular dystrophies and congenital myopathies. Patients with these conditions share characteristics such as early onset of weakness and severe hypotonia. Other organs such as the brain, eyes, and skin may be involved. Diagnosis depends largely on recognition of phenotype, muscle biopsy, and mutation analysis.
Recent findings: More than 30 genes have been associated with these diseases, most of which have only been recognized in the past decade. Increasing availability of DNA analysis has been important in decreasing delay in diagnosis.
Summary: Patients with congenital muscular dystrophy or congenital myopathy are at high risk of complications including restrictive lung disease, orthopedic deformities, seizures, cardiomyopathy, and malignant hyperthermia. Life expectancy varies with the severity of complications. Having an accurate and specific diagnosis allows the neurologist to carry out anticipatory guidance and appropriate monitoring. New hope exists for experimental treatments for congenital muscular dystrophy and congenital myopathy as our understanding of pathogenesis evolves.
Figures
References
-
- Tome FM,, Evangelista T,, Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci III 1994; 317 (4): 351–357. - PubMed
-
- Clement EM,, Feng L,, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001-2008. Neuromuscul Disord 2012; 22 (6): 522–527. - PubMed
-
- Peat RA,, Smith JM,, Compton AG, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology 2008; 71 (5): 312–321. - PubMed
-
- Okada M,, Kawahara G,, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology 2007; 69 (10): 1035–1042. - PubMed
-
- Geranmayeh F,, Clement E,, Feng LH, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord 2010; 20 (4): 241–250. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous