

Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias
- PMID: 24307138
- PMCID: PMC3943639
- DOI: 10.1007/s12311-013-0539-y
Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias
Abstract
Intensive scientific research devoted in the recent years to understand the molecular mechanisms or neurodegeneration in spinocerebellar ataxias (SCAs) are identifying new pathways and targets providing new insights and a better understanding of the molecular pathogenesis in these diseases. In this consensus manuscript, the authors discuss their current views on the identified molecular processes causing or modulating the neurodegenerative phenotype in spinocerebellar ataxias with the common opinion of translating the new knowledge acquired into candidate targets for therapy. The following topics are discussed: transcription dysregulation, protein aggregation, autophagy, ion channels, the role of mitochondria, RNA toxicity, modulators of neurodegeneration and current therapeutic approaches. Overall point of consensus includes the common vision of neurodegeneration in SCAs as a multifactorial, progressive and reversible process, at least in early stages. Specific points of consensus include the role of the dysregulation of protein folding, transcription, bioenergetics, calcium handling and eventual cell death with apoptotic features of neurons during SCA disease progression. Unresolved questions include how the dysregulation of these pathways triggers the onset of symptoms and mediates disease progression since this understanding may allow effective treatments of SCAs within the window of reversibility to prevent early neuronal damage. Common opinions also include the need for clinical detection of early neuronal dysfunction, for more basic research to decipher the early neurodegenerative process in SCAs in order to give rise to new concepts for treatment strategies and for the translation of the results to preclinical studies and, thereafter, in clinical practice.
Figures
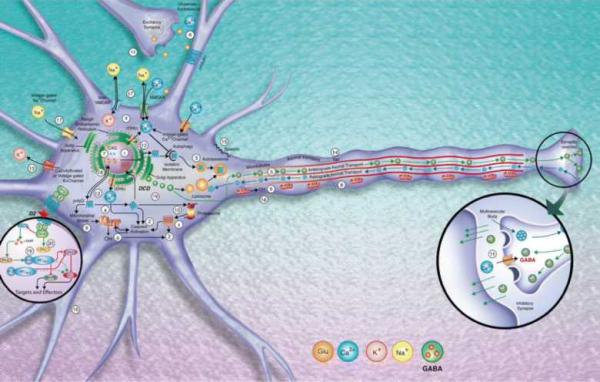
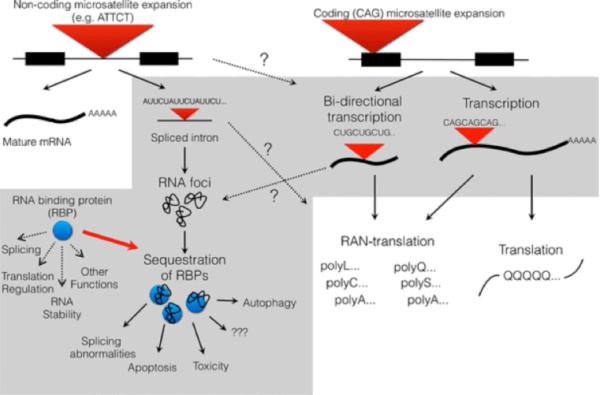
References
-
- Orr H, Chung M-y, Banfi S, Kwiatkowski TJ, Jr, Servadio A, Beaudet AL, et al. Expansion of an unstable trinucleotide (CAG) repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–6. - PubMed
-
- Dohlinger S, Hauser TK, Borkert J, Luft AR, Schulz JB. Magnetic resonance imaging in spinocerebellar ataxias. Cerebellum. 2008;7:204–14. - PubMed
-
- Reetz K, Costa AS, Mirzazade S, Lehmann A, Juzek A, Rakowicz M, et al. Genotype-specific patterns of atrophy progression are more sensitive than clinical decline in SCA1, SCA3 and SCA6. Brain. 2013;136:905–17. - PubMed
-
- Manto MU. The wide spectrum of spinocerebellar ataxias (SCAs) Cerebellum. 2005;4:2–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
