

De novo prion aggregates trigger autophagy in skeletal muscle
- PMID: 24307586
- PMCID: PMC3911572
- DOI: 10.1128/JVI.02279-13
De novo prion aggregates trigger autophagy in skeletal muscle
Abstract
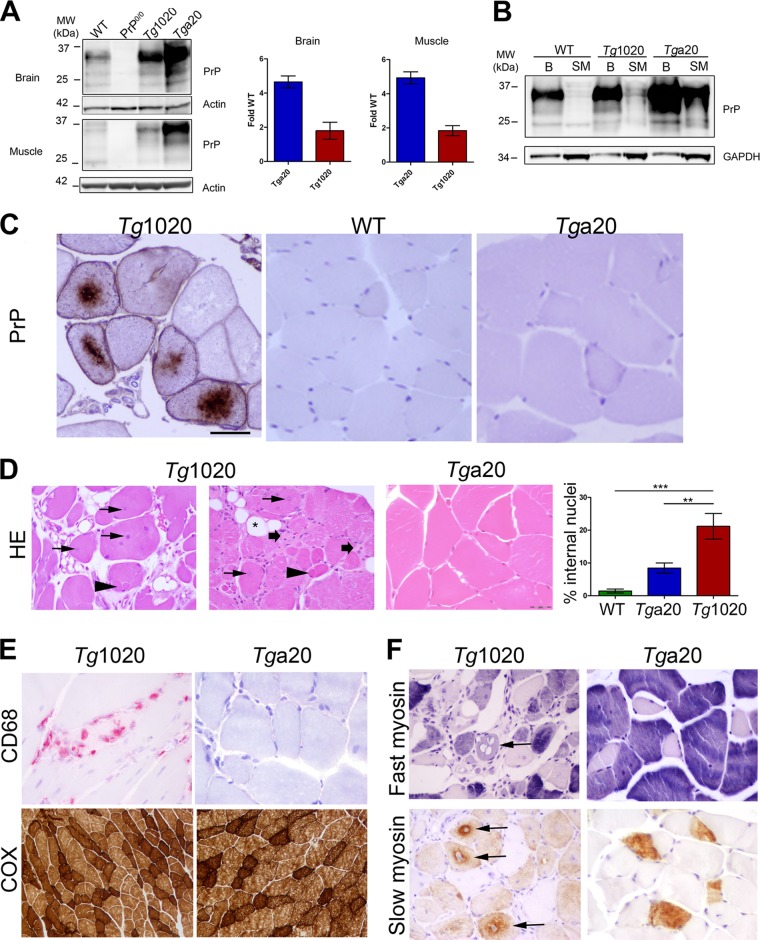
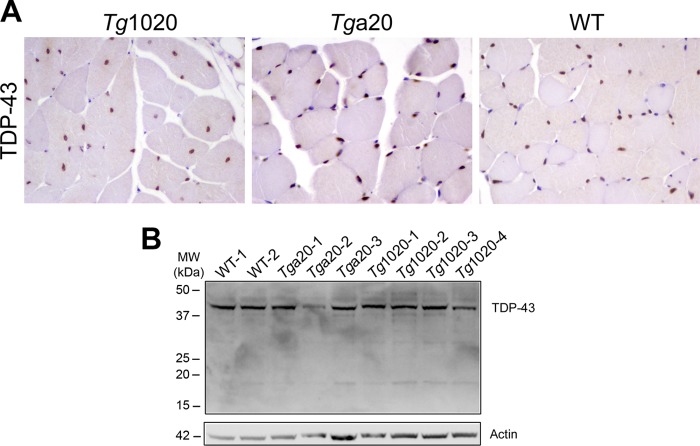
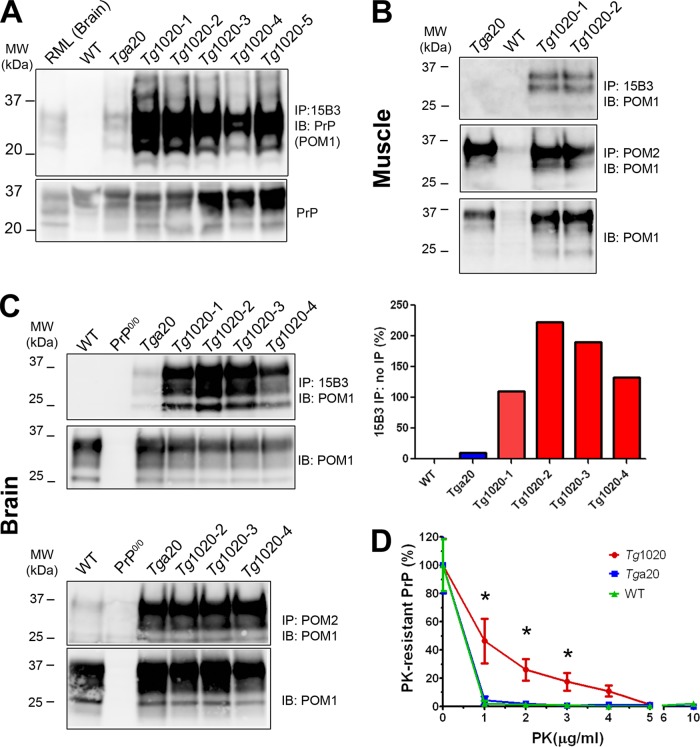
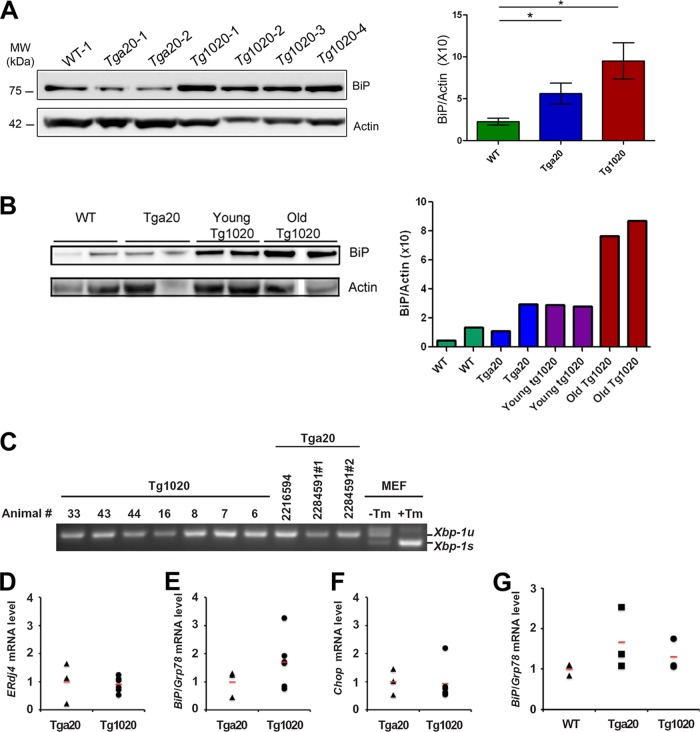
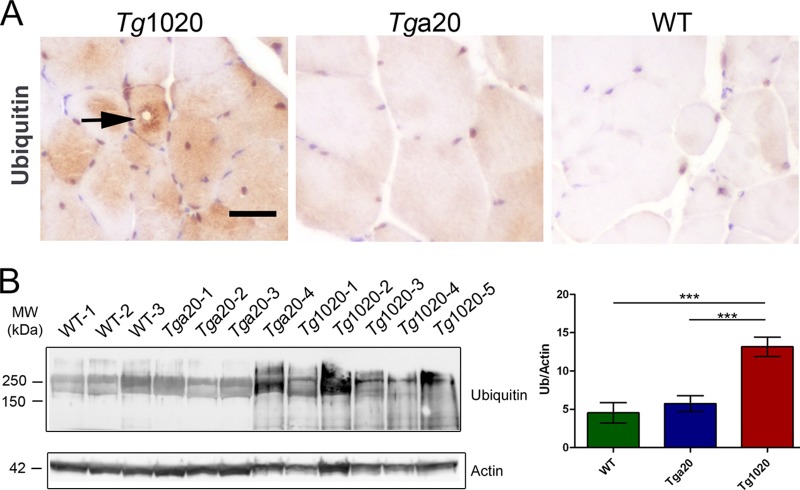
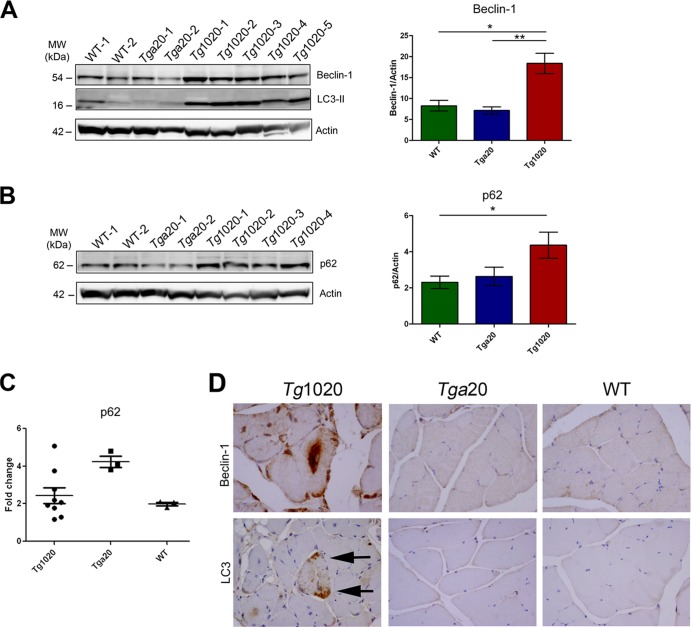
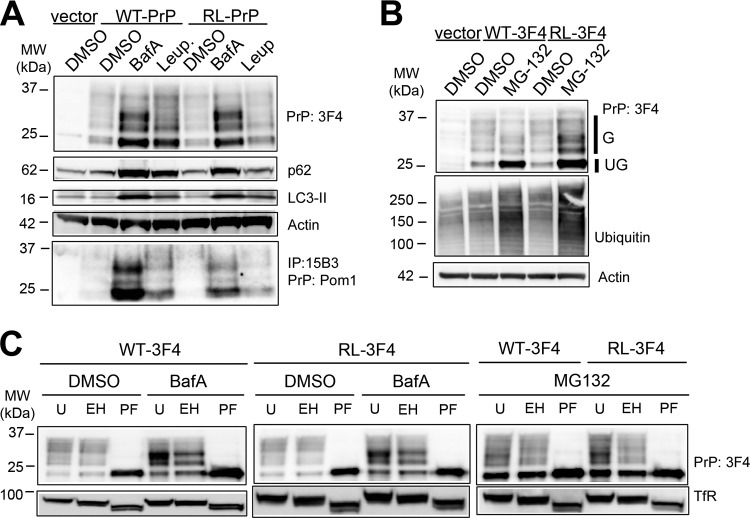
In certain sporadic, familial, and infectious prion diseases, the prion protein misfolds and aggregates in skeletal muscle in addition to the brain and spinal cord. In myocytes, prion aggregates accumulate intracellularly, yet little is known about clearance pathways. Here we investigated the clearance of prion aggregates in muscle of transgenic mice that develop prion disease de novo. In addition to neurodegeneration, aged mice developed a degenerative myopathy, with scattered myocytes containing ubiquitinated, intracellular prion inclusions that were adjacent to myocytes lacking inclusions. Myocytes also showed elevated levels of the endoplasmic reticulum chaperone Grp78/BiP, suggestive of impaired protein degradation and endoplasmic reticulum stress. Additionally, autophagy was induced, as indicated by increased levels of beclin-1 and LC3-II. In C2C12 myoblasts, inhibition of autophagosome maturation or lysosomal degradation led to enhanced prion aggregation, consistent with a role for autophagy in prion aggregate clearance. Taken together, these findings suggest that the induction of autophagy may be a central strategy for prion aggregate clearance in myocytes. IMPORTANCE In prion diseases, the prion protein misfolds and aggregates in the central nervous system and sometimes in other organs, including muscle, yet the cellular pathways of prion aggregate clearance are unclear. Here we investigated the clearance of prion aggregates in the muscle of a transgenic mouse model that develops profound muscle degeneration. We found that endoplasmic reticulum stress pathways were activated and that autophagy was induced. Blocking of autophagic degradation in cell culture models led to an accumulation of aggregated prion protein. Collectively, these findings suggest that autophagy has an instrumental role in prion protein clearance.
Figures
Similar articles
-
Autophagy, prion infection and their mutual interactions.Curr Issues Mol Biol. 2010;12(2):87-97. Epub 2009 Sep 18. Curr Issues Mol Biol. 2010. PMID: 19767652 Review.
-
Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers.Am J Pathol. 2010 Sep;177(3):1377-87. doi: 10.2353/ajpath.2010.100050. Epub 2010 Jul 8. Am J Pathol. 2010. PMID: 20616343 Free PMC article.
-
Reticulon 3 attenuates the clearance of cytosolic prion aggregates via inhibiting autophagy.Autophagy. 2011 Feb;7(2):205-16. doi: 10.4161/auto.7.2.14197. Epub 2011 Feb 1. Autophagy. 2011. PMID: 21119307
-
Muscle-dominant wild-type TDP-43 expression induces myopathological changes featuring tubular aggregates and TDP-43-positive inclusions.Exp Neurol. 2018 Nov;309:169-180. doi: 10.1016/j.expneurol.2018.08.006. Epub 2018 Aug 18. Exp Neurol. 2018. PMID: 30130494
-
An Update on Autophagy in Prion Diseases.Front Bioeng Biotechnol. 2020 Aug 27;8:975. doi: 10.3389/fbioe.2020.00975. eCollection 2020. Front Bioeng Biotechnol. 2020. PMID: 32984276 Free PMC article. Review.
Cited by
-
Excess PrPC inhibits muscle cell differentiation via miRNA-enhanced liquid-liquid phase separation implicated in myopathy.Nat Commun. 2023 Dec 8;14(1):8131. doi: 10.1038/s41467-023-43826-7. Nat Commun. 2023. PMID: 38065962 Free PMC article.
-
NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson's disease.Sci Rep. 2016 Mar 10;6:22866. doi: 10.1038/srep22866. Sci Rep. 2016. PMID: 26960433 Free PMC article.
-
Autophagy protects against de novo formation of the [PSI+] prion in yeast.Mol Biol Cell. 2015 Dec 15;26(25):4541-51. doi: 10.1091/mbc.E15-08-0548. Epub 2015 Oct 21. Mol Biol Cell. 2015. PMID: 26490118 Free PMC article.
-
The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective.Trends Neurosci. 2016 Apr;39(4):221-234. doi: 10.1016/j.tins.2016.02.002. Epub 2016 Mar 9. Trends Neurosci. 2016. PMID: 26968346 Free PMC article. Review.
-
Prion formation, but not clearance, is supported by protein misfolding cyclic amplification.Prion. 2014;8(6):415-20. doi: 10.4161/19336896.2014.983759. Prion. 2014. PMID: 25482601 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
