

The role of cell signalling in the crosstalk between autophagy and apoptosis
- PMID: 24308968
- PMCID: PMC4054685
- DOI: 10.1016/j.cellsig.2013.11.028
The role of cell signalling in the crosstalk between autophagy and apoptosis
Abstract
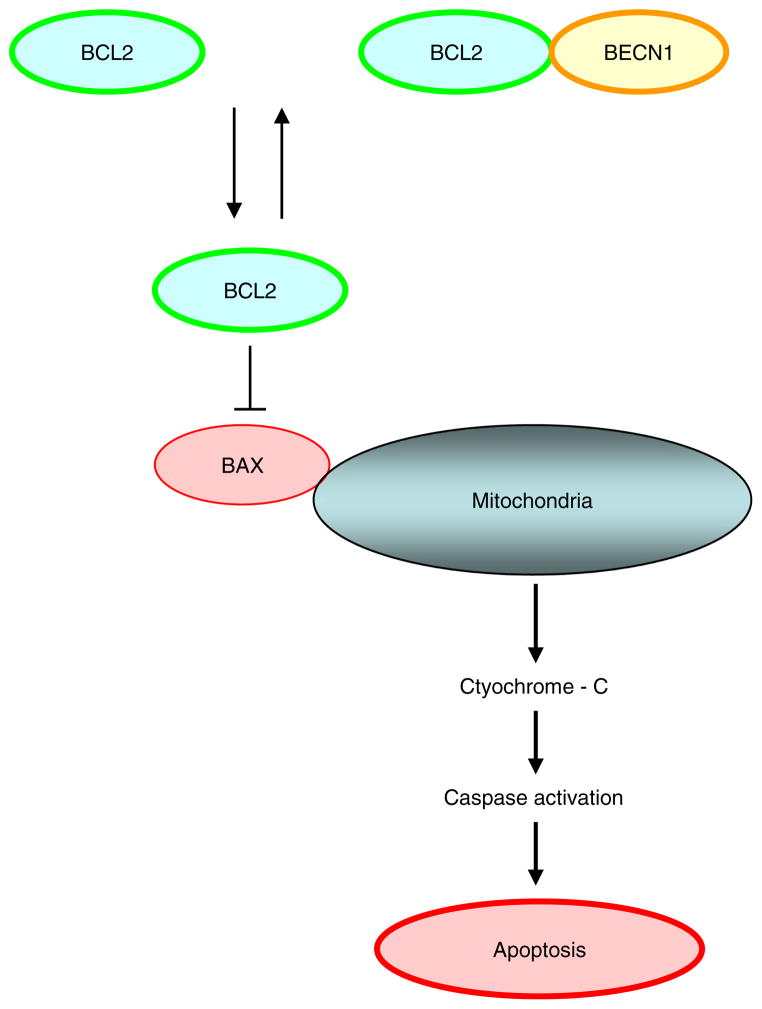
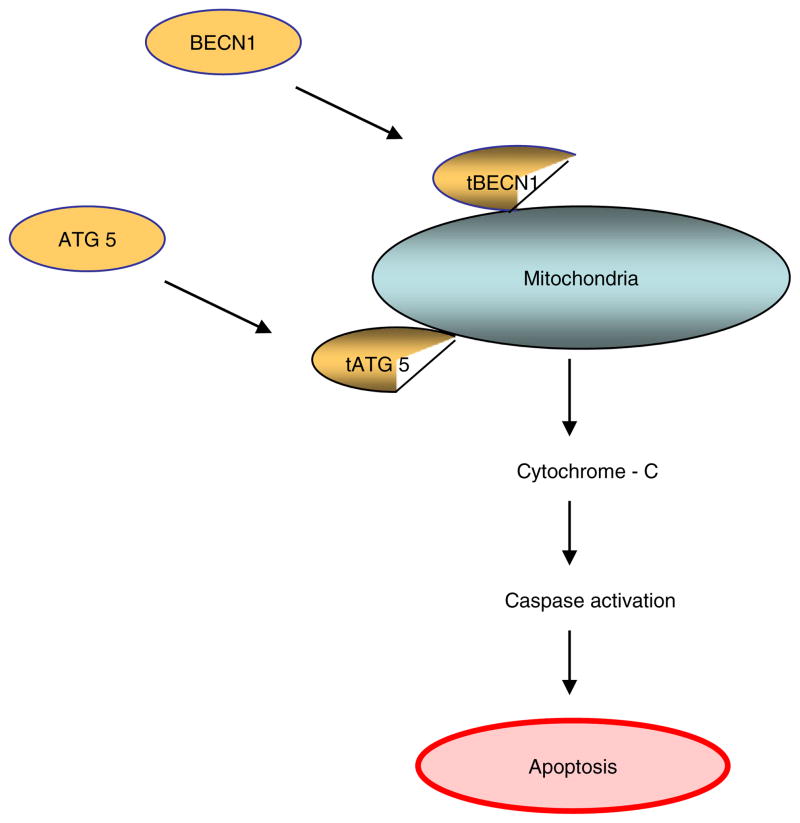
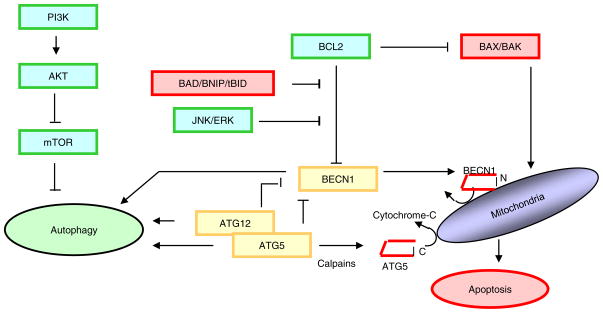
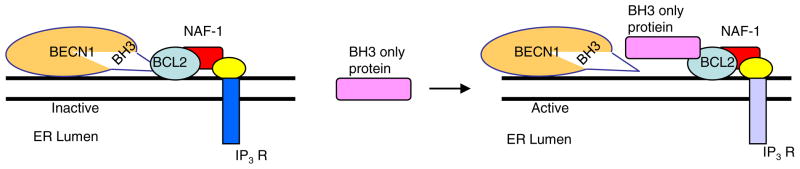
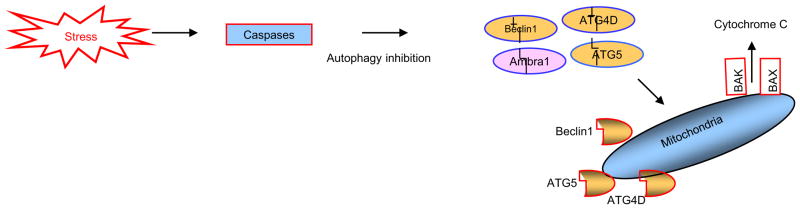
Not surprisingly, the death of a cell is a complex and well controlled process. For several decades, apoptosis, the first genetically programmed death process to be identified has taken centre stage as the principal mechanism of programmed cell death (type I cell death) in mammalian tissues. Apoptosis has been extensively studied and its contribution to the pathogenesis of disease well documented. However, apoptosis does not function alone in determining the fate of a cell. More recently, autophagy, a process in which de novo formed membrane enclosed vesicles engulf and consume cellular components, has been shown to engage in complex interplay with apoptosis. As a result, cell death has been subdivided into the categories apoptosis (Type I), autophagic cell death (Type II), and necrosis (Type III). The boundary between Type I and II cell death is not completely clear and as we will discuss in this review and perhaps a discrete difference does not exist, due to intrinsic factors among different cell types and crosstalk among organelles within each cell type. Apoptosis may begin with autophagy and autophagy can often end with apoptosis, inhibition or a blockade of caspase activity may lead a cell to default into Type II cell death from Type I.
Keywords: Apoptosis; Autophagosome; Autophagy; Caspase; Sorafenib.
Published by Elsevier Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
