

Functional interpretation of non-coding sequence variation: concepts and challenges
- PMID: 24311363
- PMCID: PMC3992842
- DOI: 10.1002/bies.201300126
Functional interpretation of non-coding sequence variation: concepts and challenges
Abstract
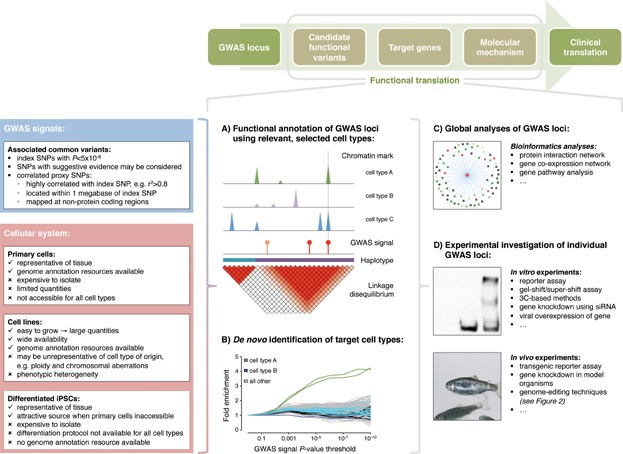
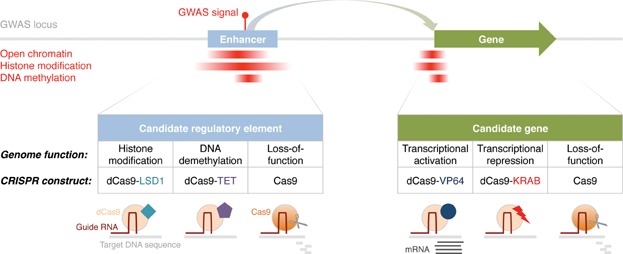
Understanding the functional mechanisms underlying genetic signals associated with complex traits and common diseases, such as cancer, diabetes and Alzheimer's disease, is a formidable challenge. Many genetic signals discovered through genome-wide association studies map to non-protein coding sequences, where their molecular consequences are difficult to evaluate. This article summarizes concepts for the systematic interpretation of non-coding genetic signals using genome annotation data sets in different cellular systems. We outline strategies for the global analysis of multiple association intervals and the in-depth molecular investigation of individual intervals. We highlight experimental techniques to validate candidate (potential causal) regulatory variants, with a focus on novel genome-editing techniques including CRISPR/Cas9. These approaches are also applicable to low-frequency and rare variants, which have become increasingly important in genomic studies of complex traits and diseases. There is a pressing need to translate genetic signals into biological mechanisms, leading to prognostic, diagnostic and therapeutic advances.
Keywords: GWAS; chromatin; complex traits; gene regulation; genome editing; regulatory variants.
© 2014 The Authors. Bioessays published by WILEY Periodicals, Inc.
Figures
References
-
- Mardis ER. A decade's perspective on DNA sequencing technology. Nature. 2011;470:198–203. - PubMed
-
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. - PubMed
-
- Donnelly P. Progress and challenges in genome-wide association studies in humans. Nature. 2008;456:728–31. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
