

Genome-wide comparison of cowpox viruses reveals a new clade related to Variola virus
- PMID: 24312452
- PMCID: PMC3848979
- DOI: 10.1371/journal.pone.0079953
Genome-wide comparison of cowpox viruses reveals a new clade related to Variola virus
Abstract
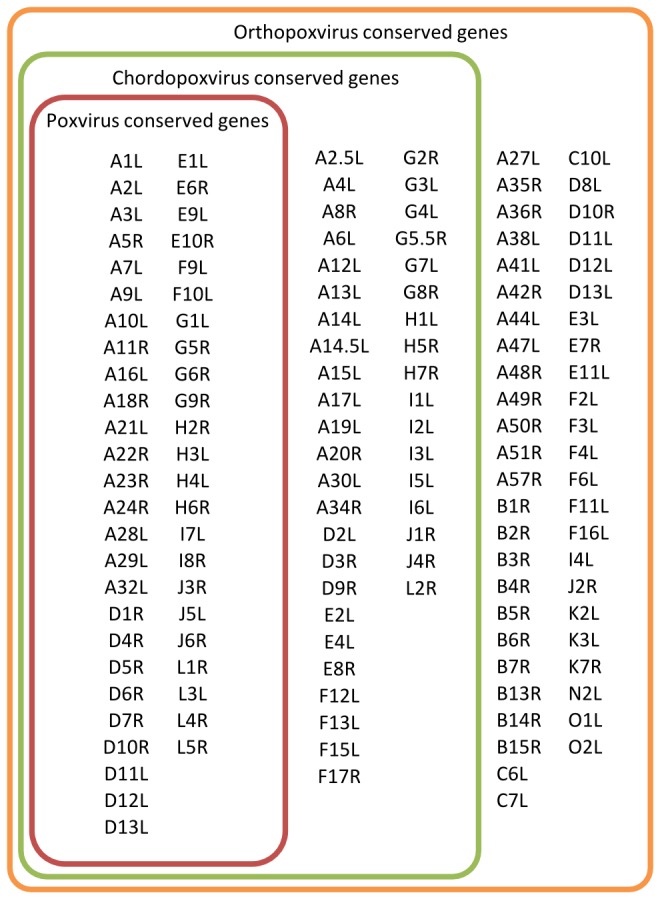
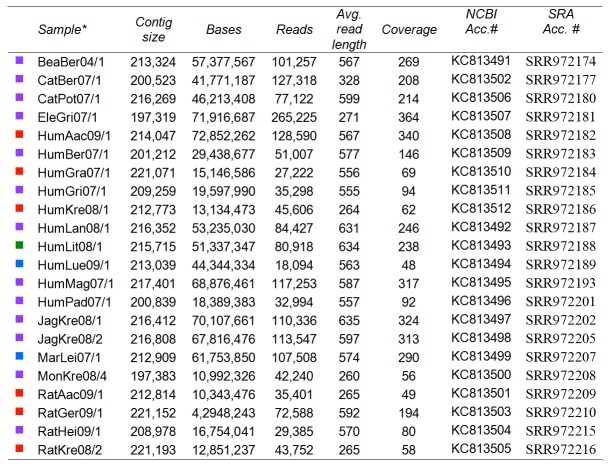
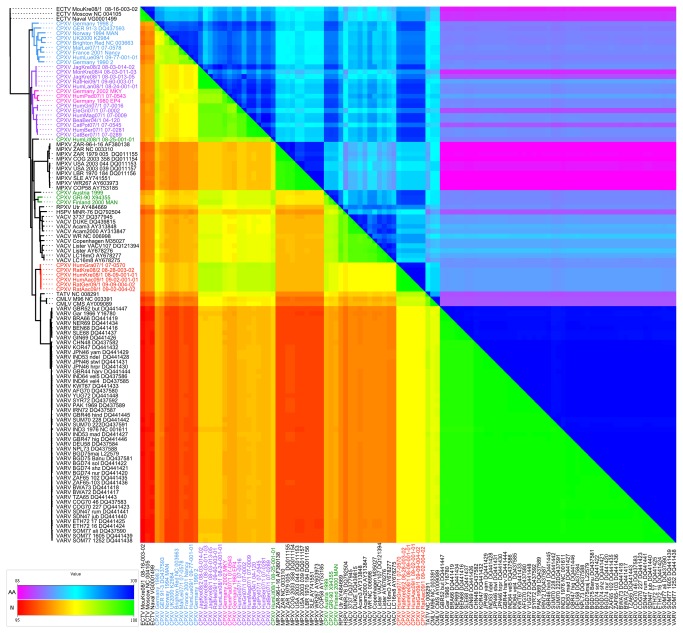
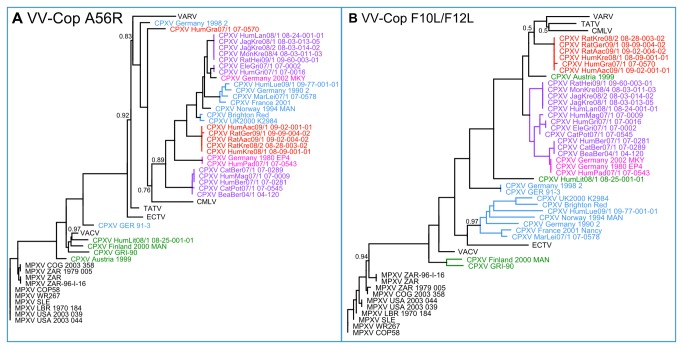
Zoonotic infections caused by several orthopoxviruses (OPV) like monkeypox virus or vaccinia virus have a significant impact on human health. In Europe, the number of diagnosed infections with cowpox viruses (CPXV) is increasing in animals as well as in humans. CPXV used to be enzootic in cattle; however, such infections were not being diagnosed over the last decades. Instead, individual cases of cowpox are being found in cats or exotic zoo animals that transmit the infection to humans. Both animals and humans reveal local exanthema on arms and legs or on the face. Although cowpox is generally regarded as a self-limiting disease, immunosuppressed patients can develop a lethal systemic disease resembling smallpox. To date, only limited information on the complex and, compared to other OPV, sparsely conserved CPXV genomes is available. Since CPXV displays the widest host range of all OPV known, it seems important to comprehend the genetic repertoire of CPXV which in turn may help elucidate specific mechanisms of CPXV pathogenesis and origin. Therefore, 22 genomes of independent CPXV strains from clinical cases, involving ten humans, four rats, two cats, two jaguarundis, one beaver, one elephant, one marah and one mongoose, were sequenced by using massive parallel pyrosequencing. The extensive phylogenetic analysis showed that the CPXV strains sequenced clearly cluster into several distinct clades, some of which are closely related to Vaccinia viruses while others represent different clades in a CPXV cluster. Particularly one CPXV clade is more closely related to Camelpox virus, Taterapox virus and Variola virus than to any other known OPV. These results support and extend recent data from other groups who postulate that CPXV does not form a monophyletic clade and should be divided into multiple lineages.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
