

Epigenetic targets for reversing immune defects caused by alcohol exposure
- PMID: 24313169
- PMCID: PMC3860427
Epigenetic targets for reversing immune defects caused by alcohol exposure
Abstract
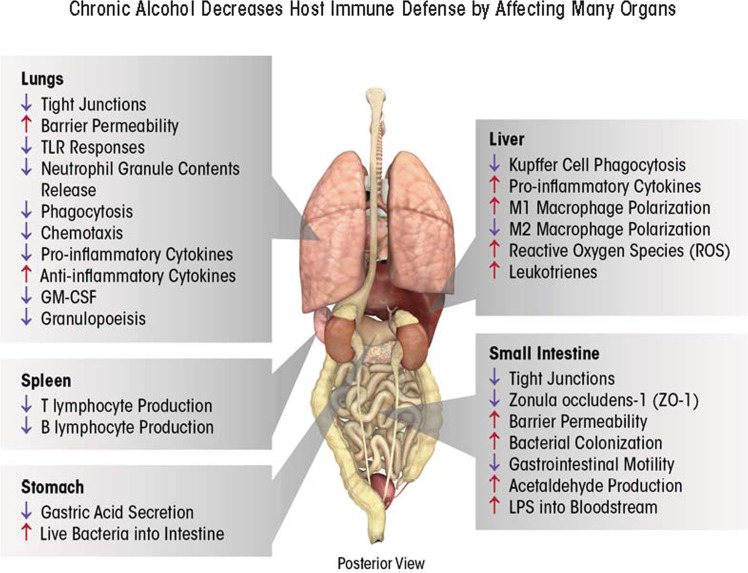
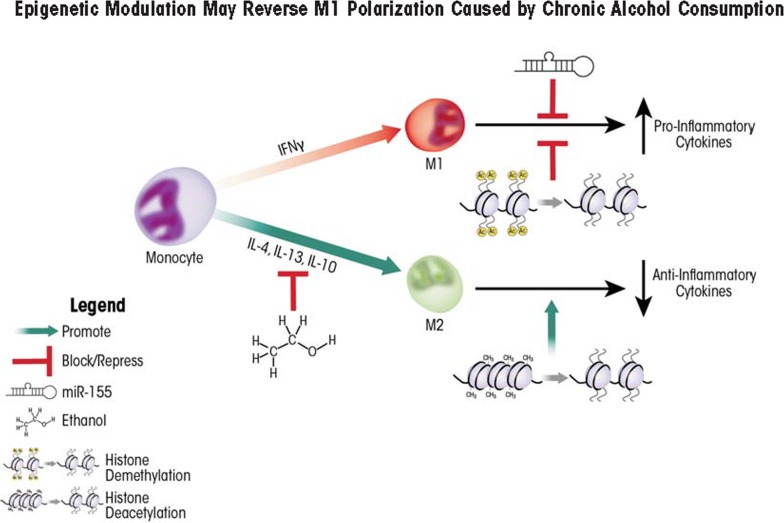
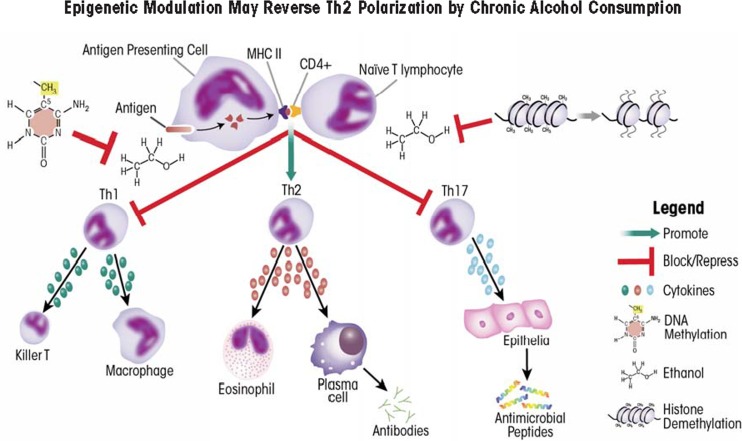
Alcohol consumption alters factors that modify gene expression without changing the DNA code (i.e., epigenetic modulators) in many organ systems, including the immune system. Alcohol enhances the risk for developing several serious medical conditions related to immune system dysfunction, including acute respiratory distress syndrome (ARDS), liver cancer, and alcoholic liver disease (ALD). Binge and chronic drinking also render patients more susceptible to many infectious pathogens and advance the progression of HIV infection by weakening both innate and adaptive immunity. Epigenetic mechanisms play a pivotal role in these processes. For example, alcohol-induced epigenetic variations alter the developmental pathways of several types of immune cells (e.g., granulocytes, macrophages, and T-lymphocytes) and through these and other mechanisms promote exaggerated inflammatory responses. In addition, epigenetic mechanisms may underlie alcohol's ability to interfere with the barrier functions of the gut and respiratory systems, which also contribute to the heightened risk of infections. Better understanding of alcohol's effects on these epigenetic processes may help researchers identify new targets for the development of novel medications to prevent or ameliorate alcohol's detrimental effects on the immune system.
Figures
References
-
- Albano E. Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Molecular Aspects of Medicine. 2008;29(1–2):9–16. - PubMed
-
- Atkinson KJ, Rao RK. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. American Journal of Physiology. Gastrointestinal and Liver Physiology. 2001;280(6):G1280–G1288. - PubMed
-
- Bagby GJ, Zhang P, Stoltz DA, Nelson S. Suppression of the granulocyte colony-stimulating factor response to Escherichia coli challenge by alcohol intoxication. Alcoholism: Clinical and Experimental Research. 1998;22(8):1740–1745. - PubMed
-
- Baliunas D, Rehm J, Irving H, Shuper P. Alcohol consumption and risk of incident human immunodeficiency virus infection: A meta-analysis. International Journal of Public Health. 2010;55(3):159–166. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
