

Pathophysiology of X-linked adrenoleukodystrophy
- PMID: 24316281
- PMCID: PMC3988840
- DOI: 10.1016/j.biochi.2013.11.023
Pathophysiology of X-linked adrenoleukodystrophy
Abstract
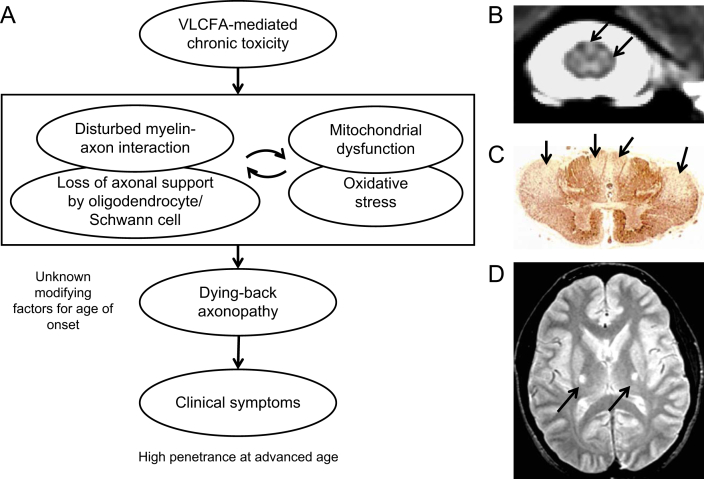
Currently the molecular basis for the clinical heterogeneity of X-linked adrenoleukodystrophy (X-ALD) is poorly understood. The genetic bases for all different phenotypic variants of X-ALD are mutations in the gene encoding the peroxisomal ATP-binding cassette (ABC) transporter, ABCD1 (formerly adrenoleukodystrophy protein, ALDP). ABCD1 transports CoA-activated very long-chain fatty acids from the cytosol into the peroxisome for degradation. The phenotypic variability is remarkable ranging from cerebral inflammatory demyelination of childhood onset, leading to death within a few years, to adults remaining pre-symptomatic through more than five decades. There is no general genotype-phenotype correlation in X-ALD. The default manifestation of mutations in ABCD1 is adrenomyeloneuropathy, a slowly progressive dying-back axonopathy affecting both ascending and descending spinal cord tracts as well as in some cases, a peripheral neuropathy. In about 60% of male X-ALD patients, either in childhood (35-40%) or in adulthood (20%), an initial, clinically silent, myelin destabilization results in conversion to a devastating, rapidly progressive form of cerebral inflammatory demyelination. Here, ABCD1 remains a susceptibility gene, necessary but not sufficient for inflammatory demyelination to occur. Although the accumulation of very long-chain fatty acids appears to be essential for the pathomechanism of all phenotypes, the molecular mechanisms underlying these phenotypes are fundamentally different. Cell autonomous processes such as oxidative stress and energy shortage in axons as well as non-cell autonomous processes involving axon-glial interactions seem pertinent to the dying-back axonopathy. Various dynamic mechanisms may underlie the initiation of inflammation, the altered immune reactivity, the propagation of inflammation, as well as the mechanisms leading to the arrest of inflammation after hematopoietic stem cell transplantation. An improved understanding of the molecular mechanisms involved in these events is required for the development of urgently needed therapeutics.
Keywords: ABC transporter; Axonopathy; Demyelination; Inflammation; Leukodystrophy; Peroxisome.
Copyright © 2013 The Authors. Published by Elsevier Masson SAS.. All rights reserved.
Figures

References
-
- Bezman L., Moser H.W. Incidence of X-linked adrenoleukodystrophy and the relative frequency of its phenotypes. Am. J. Med. Genet. 1998;76:415–419. - PubMed
-
- Kemp S., Berger J., Aubourg P. X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim. Biophys. Acta. 2012;1822:1465–1474. - PubMed
-
- Moser H.W., Smith K.D., Watkins P.A., Powers J., Moser A.B. X-linked Adrenoleukodystrophy. In: Scriver R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic & Molecular Bases of Inherited Disease. eighth ed. McGraw-Hill Book Co.; New York: 2001. pp. 3257–3301.
-
- Peters C., Charnas L.R., Tan Y., Ziegler R.S., Shapiro E.G., DeFor T., Grewal S.S., Orchard P.J., Abel S.L., Goldman A.I., Ramsay N.K., Dusenbery K.E., Loes D.J., Lockman L.A., Kato S., Aubourg P.R., Moser H.W., Krivit W. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881–888. - PubMed
-
- Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., Mahlaoui N., Kiermer V., Mittelstaedt D., Bellesme C., Lahlou N., Lefrere F., Blanche S., Audit M., Payen E., Leboulch P., l'Homme B., Bougneres P., Von Kalle C., Fischer A., Cavazzana-Calvo M., Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
