

Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification
- PMID: 24317252
- PMCID: PMC4018771
- DOI: 10.1038/nmeth.2762
Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification
Abstract
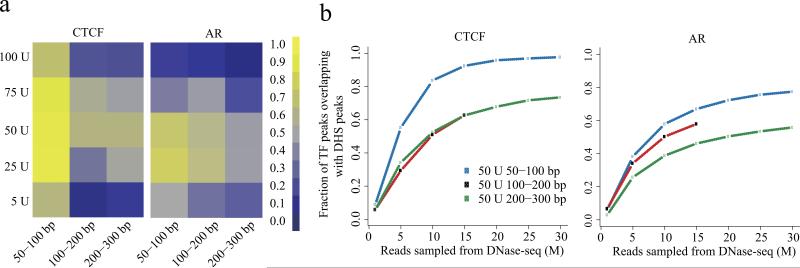
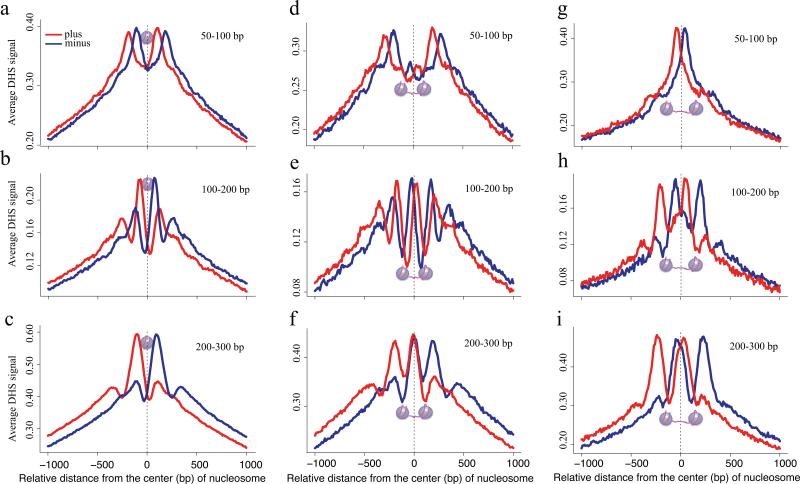
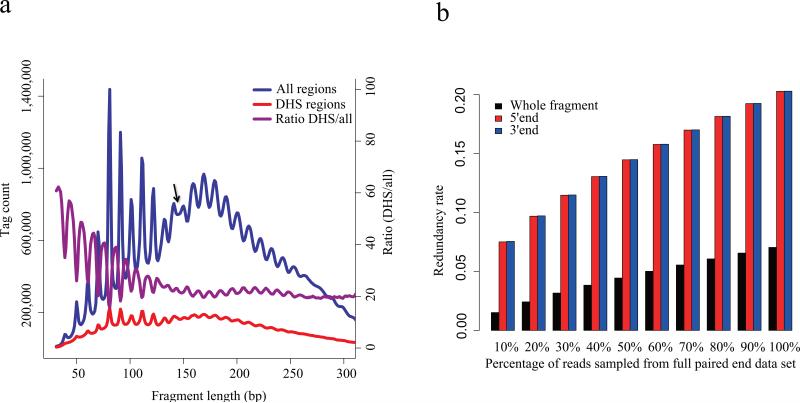
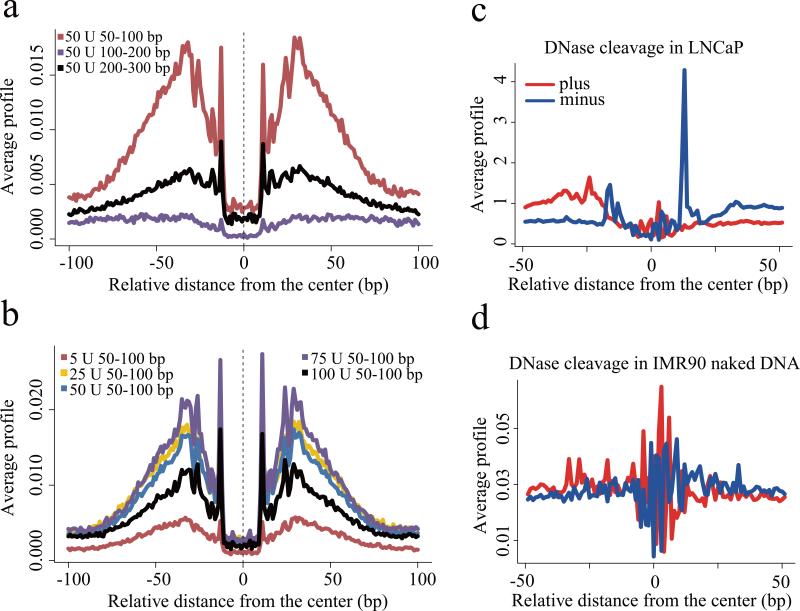
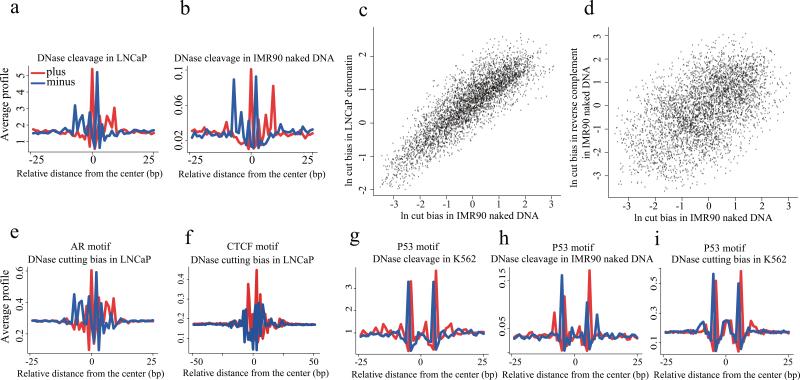
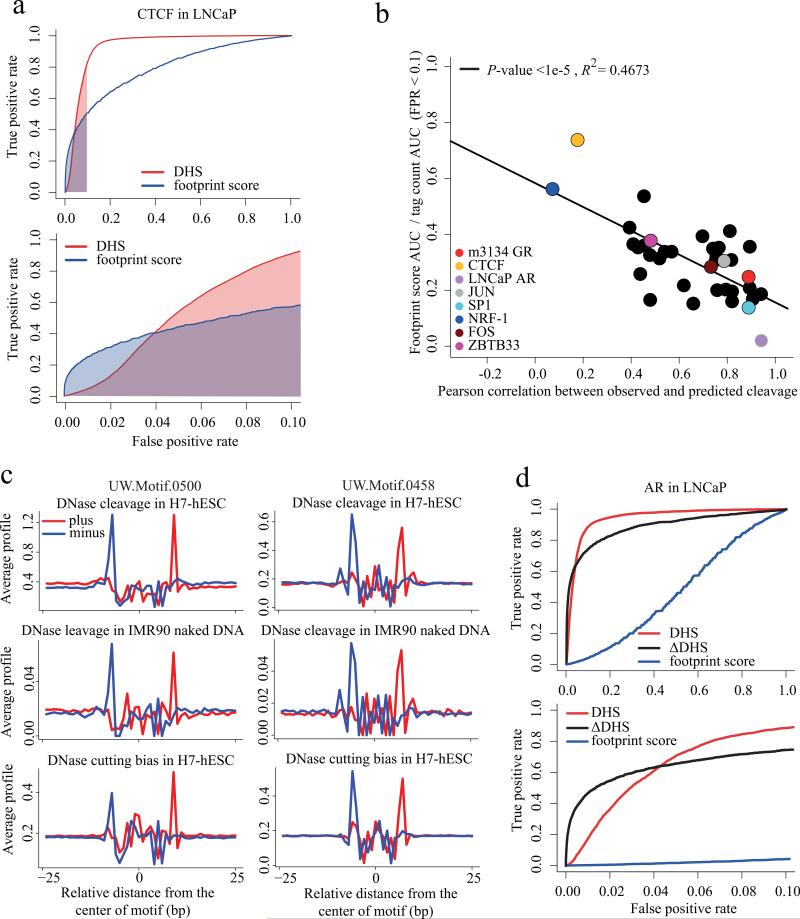
Sequencing of DNase I hypersensitive sites (DNase-seq) is a powerful technique for identifying cis-regulatory elements across the genome. We studied the key experimental parameters to optimize performance of DNase-seq. Sequencing short fragments of 50-100 base pairs (bp) that accumulate in long internucleosome linker regions was more efficient for identifying transcription factor binding sites compared to sequencing longer fragments. We also assessed the potential of DNase-seq to predict transcription factor occupancy via generation of nucleotide-resolution transcription factor footprints. In modeling the sequence-specific DNase I cutting bias, we found a strong effect that varied over more than two orders of magnitude. This indicates that the nucleotide-resolution cleavage patterns at many transcription factor binding sites are derived from intrinsic DNase I cleavage bias rather than from specific protein-DNA interactions. In contrast, quantitative comparison of DNase I hypersensitivity between states can predict transcription factor occupancy associated with particular biological perturbations.
Figures
Comment in
-
The genome shows its sensitive side.Nat Methods. 2014 Jan;11(1):39-40. doi: 10.1038/nmeth.2770. Nat Methods. 2014. PMID: 24378702 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
