

MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies
- PMID: 24323356
- PMCID: PMC3865476
- DOI: 10.1084/jem.20130699
MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies
Abstract
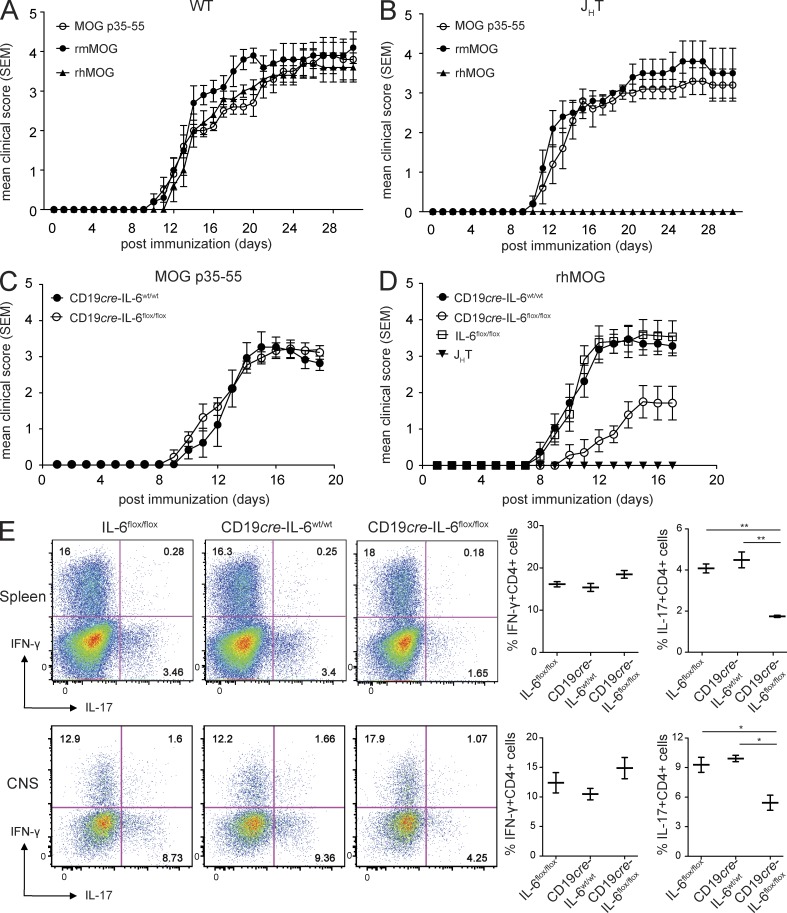
Whether B cells serve as antigen-presenting cells (APCs) for activation of pathogenic T cells in the multiple sclerosis model experimental autoimmune encephalomyelitis (EAE) is unclear. To evaluate their role as APCs, we engineered mice selectively deficient in MHC II on B cells (B-MHC II(-/-)), and to distinguish this function from antibody production, we created transgenic (Tg) mice that express the myelin oligodendrocyte glycoprotein (MOG)-specific B cell receptor (BCR; IgH(MOG-mem)) but cannot secrete antibodies. B-MHC II(-/-) mice were resistant to EAE induced by recombinant human MOG (rhMOG), a T cell- and B cell-dependent autoantigen, and exhibited diminished Th1 and Th17 responses, suggesting a role for B cell APC function. In comparison, selective B cell IL-6 deficiency reduced EAE susceptibility and Th17 responses alone. Administration of MOG-specific antibodies only partially restored EAE susceptibility in B-MHC II(-/-) mice. In the absence of antibodies, IgH(MOG-mem) mice, but not mice expressing a BCR of irrelevant specificity, were fully susceptible to acute rhMOG-induced EAE, also demonstrating the importance of BCR specificity. Spontaneous opticospinal EAE and meningeal follicle-like structures were observed in IgH(MOG-mem) mice crossed with MOG-specific TCR Tg mice. Thus, B cells provide a critical cellular function in pathogenesis of central nervous system autoimmunity independent of their humoral involvement, findings which may be relevant to B cell-targeted therapies.
Figures





References
-
- Amor S., Groome N., Linington C., Morris M.M., Dornmair K., Gardinier M.V., Matthieu J.M., Baker D. 1994. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J. Immunol. 153:4349–4356 - PubMed
-
- Bar-Or A., Calabresi P.A., Arnold D., Markowitz C., Shafer S., Kasper L.H., Waubant E., Gazda S., Fox R.J., Panzara M., et al. 2008. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann. Neurol. 63:395–400 [published erratum appears in Ann. Neurol. 2008. 63:803] 10.1002/ana.21363 - DOI - PubMed
-
- Basso A.S., Frenkel D., Quintana F.J., Costa-Pinto F.A., Petrovic-Stojkovic S., Puckett L., Monsonego A., Bar-Shir A., Engel Y., Gozin M., et al. 2008. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J. Clin. Invest. 118:1532–1543 10.1172/JCI33464 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
