

Resolution doubling in fluorescence microscopy with confocal spinning-disk image scanning microscopy
- PMID: 24324140
- PMCID: PMC3876259
- DOI: 10.1073/pnas.1315858110
Resolution doubling in fluorescence microscopy with confocal spinning-disk image scanning microscopy
Abstract
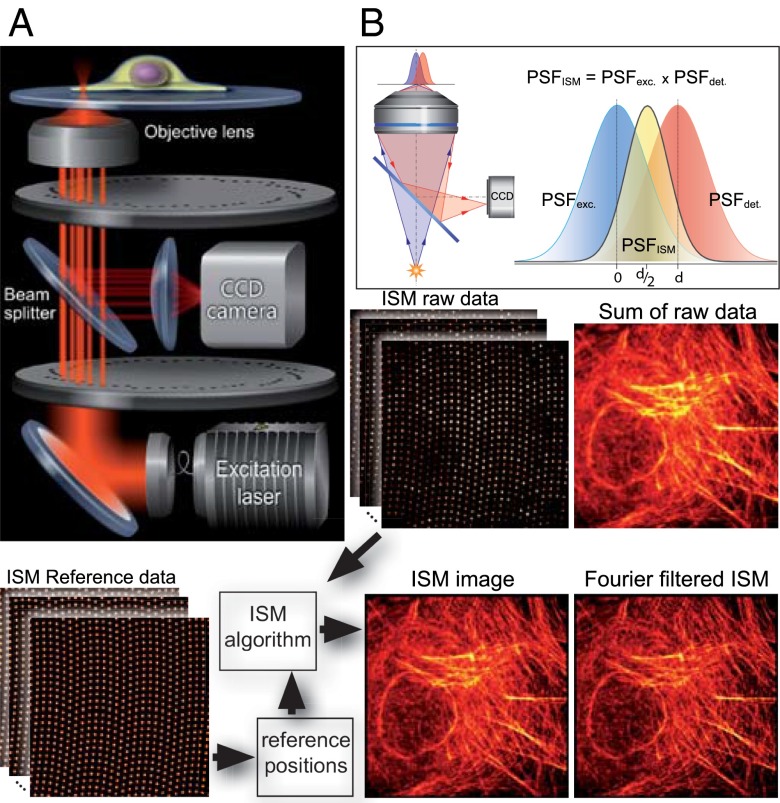
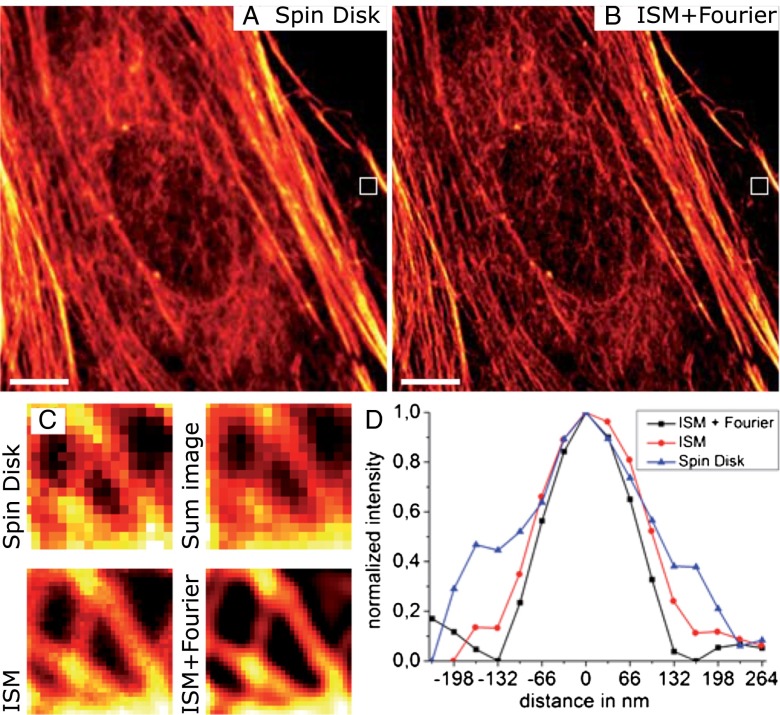
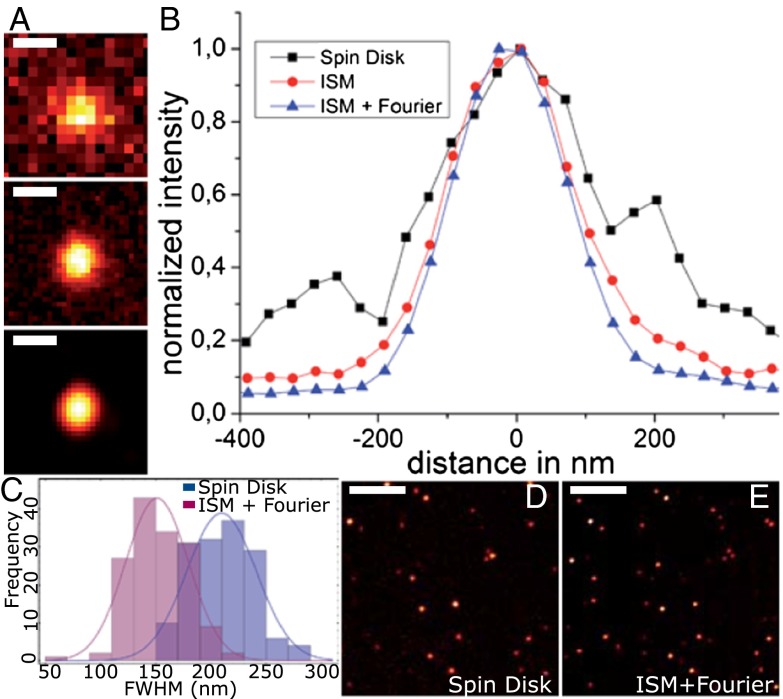
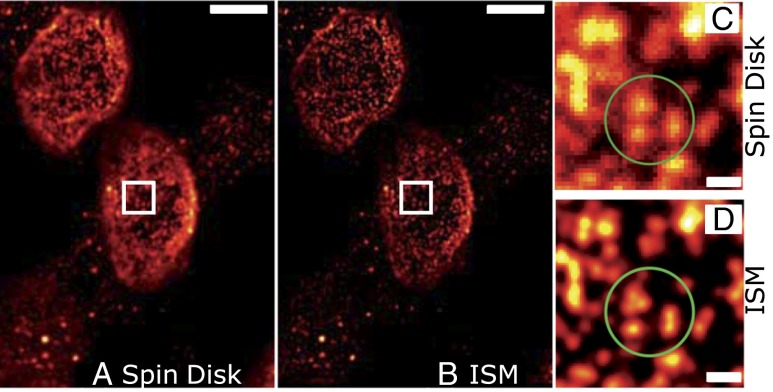
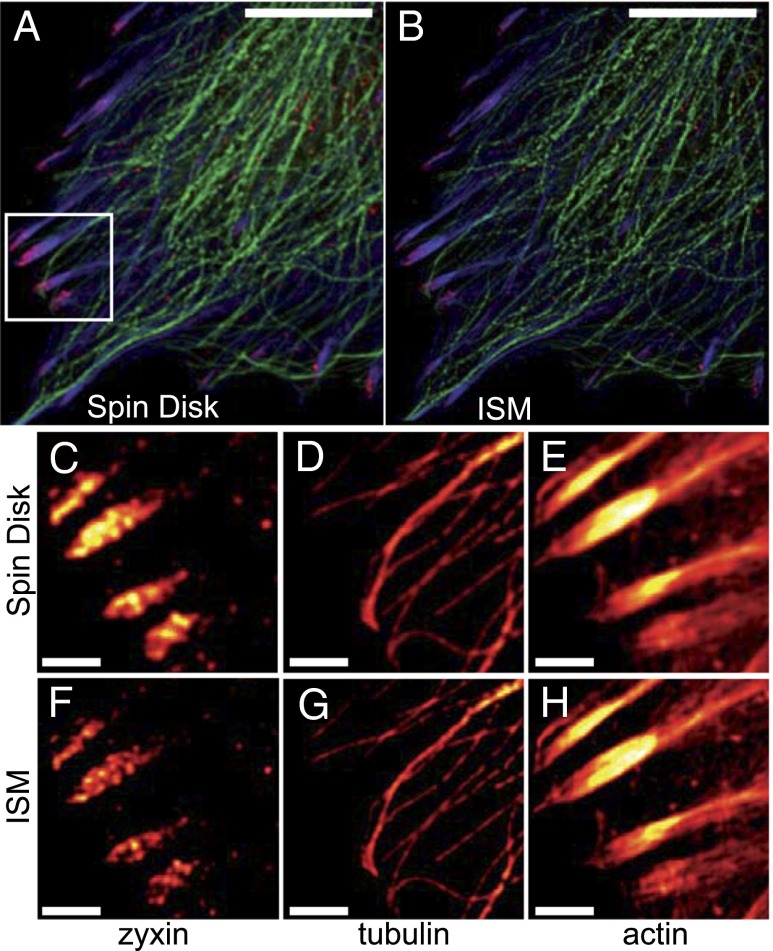
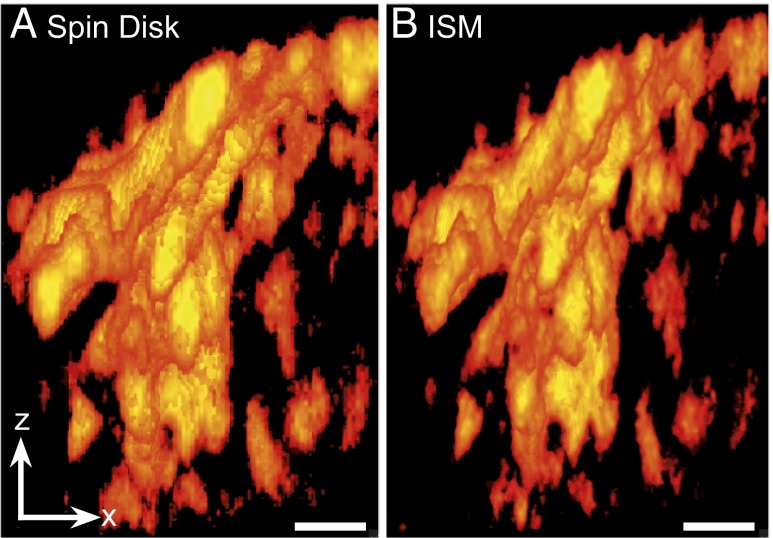
We demonstrate how a conventional confocal spinning-disk (CSD) microscope can be converted into a doubly resolving image scanning microscopy (ISM) system without changing any part of its optical or mechanical elements. Making use of the intrinsic properties of a CSD microscope, we illuminate stroboscopically, generating an array of excitation foci that are moved across the sample by varying the phase between stroboscopic excitation and rotation of the spinning disk. ISM then generates an image with nearly doubled resolution. Using conventional fluorophores, we have imaged single nuclear pore complexes in the nuclear membrane and aggregates of GFP-conjugated Tau protein in three dimensions. Multicolor ISM was shown on cytoskeletal-associated structural proteins and on 3D four-color images including MitoTracker and Hoechst staining. The simple adaptation of conventional CSD equipment allows superresolution investigations of a broad variety of cell biological questions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt Lett. 1994;19(11):780–782. - PubMed
-
- Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. - PubMed
-
- Müller CB, Enderlein J. Image scanning microscopy. Phys Rev Lett. 2010;104(19):198101. - PubMed
-
- Gustafsson MG. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc. 2000;198(Pt 2):82–87. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
