

Genetic and epigenetic control of metabolic health
- PMID: 24327950
- PMCID: PMC3854991
- DOI: 10.1016/j.molmet.2013.09.002
Genetic and epigenetic control of metabolic health
Abstract
Obesity is characterized as an excess accumulation of body fat resulting from a positive energy balance. It is the major risk factor for type 2 diabetes (T2D). The evidence for familial aggregation of obesity and its associated metabolic diseases is substantial. To date, about 150 genetic loci identified in genome-wide association studies (GWAS) are linked with obesity and T2D, each accounting for only a small proportion of the predicted heritability. However, the percentage of overall trait variance explained by these associated loci is modest (~5-10% for T2D, ~2% for BMI). The lack of powerful genetic associations suggests that heritability is not entirely attributable to gene variations. Some of the familial aggregation as well as many of the effects of environmental exposures, may reflect epigenetic processes. This review summarizes our current knowledge on the genetic basis to individual risk of obesity and T2D, and explores the potential role of epigenetic contribution.
Keywords: ADCY3, adenylate cyclase 3; AQP9, aquaporin 9; BDNF, brain-derived neurotrophic factor; CDKAL1, CDK5 regulatory subunit associated protein 1-like 1; CPEB4, cytoplasmic polyadenylation element binding protein 4; DUSP22, dual specificity phosphatase 22; DUSP8, dual specificity phosphatase 8; Epigenetics; GALNT10, UDP-N-acetyl-alpha-d-galactosamine:polypeptide N-acetylgalactosaminyltransferase 10 (GalNAc-T10); GIPR, gastric inhibitory polypeptide receptor; GNPDA2, glucosamine-6-phosphate deaminase 2; GP2, glycoprotein 2 (zymogen granule membrane); GWAS; HIPK3, homeodomain interacting protein kinase 3; IFI16, interferon, gamma-inducible protein 16; KCNQ1, potassium voltage-gated channel, KQT-like subfamily, member 1; KLHL32, kelch-like family member 32; LEPR, leptin receptor; MAP2K4, mitogen-activated protein kinase kinase 4; MAP2K5, mitogen-activated protein kinase kinase 5; MIR148A, microRNA 148a; MMP9, matrix metallopeptidase 9 (gelatinase B, 92 kDa gelatinase, 92 kDa type IV collagenase); MNDA, myeloid cell nuclear differentiation antigen; NFE2L3, nuclear factor, erythroid 2-like 3; Obesity; PACS1, phosphofurin acidic cluster sorting protein 1; PAX6, paired box gene 6; PCSK1, proprotein convertase subtilisin/kexin type 1; PGC1α, peroxisome proliferative activated receptor, gamma, coactivator 1 alpha, PM2OD1; PRKCH, protein kinase C, eta; PRKD1, protein kinase D1; PRKG1, protein kinase, cGMP-dependent, type I; Positional cloning; QPCTL, glutaminyl-peptide cyclotransferase-like; RBJ, DnaJ (Hsp40) homolog, subfamily C, member 27; RFC5, replication factor C (activator 1) 5; RMST, rhabdomyosarcoma 2 associated transcript (non-protein coding); SEC16B, SEC16 homolog B; TFAP2B, transcription factor AP-2 beta (activating enhancer binding protein 2 beta); TNNI3, troponin I type 3 (cardiac); TNNT1, troponin T type 1 (skeletal, slow); Type 2 diabetes.
Figures


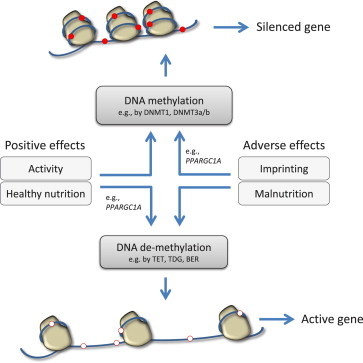
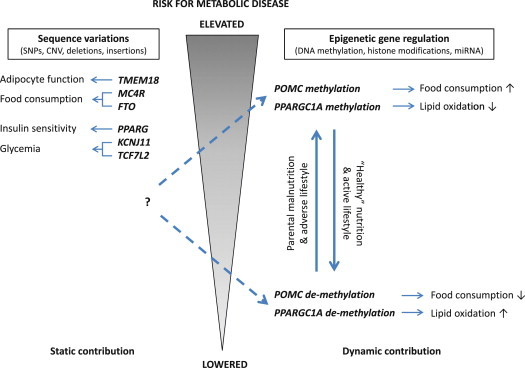
References
-
- Wynne K., Stanley S., McGowan B., Bloom S. Appetite control. Journal of Endocrinology. 2005;184:291–318. - PubMed
-
- Berthoud H.R. Multiple neural systems controlling food intake and body weight. Neuroscience & Biobehavioral Reviews. 2002;26:393–428. - PubMed
-
- López M., Tovar S., Vázquez M.J., Williams L.M., Diéguez C. Peripheral tissue-brain interactions in the regulation of food intake. Proceedings of the Nutrition Society. 2007;66:131–155. - PubMed
-
- Lam T.K., Schwartz G.J., Rossetti L. Hypothalamic sensing of fatty acids. Nature Neuroscience. 2005;8:579–584. - PubMed
-
- Blouet C., Schwartz G.J. Hypothalamic nutrient sensing in the control of energy homeostasis. Behavioural Brain Research. 2010;209:1–12. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
