

Review
doi: 10.1021/cr400477t.
Epub 2013 Dec 13.
Lanthanide probes for bioresponsive imaging
Affiliations
- PMID: 24328202
- PMCID: PMC3999228
- DOI: 10.1021/cr400477t
Item in Clipboard
Review
Lanthanide probes for bioresponsive imaging
Chem Rev.
.
No abstract available
Figures

(A) The antenna effect.,,, The general architecture of emissive lanthanide complexes consists of the metal center surrounded by a chelate and equipped with a sensitizer or antenna. The chelate serves to prevent the release of free lanthanides into the biological systems and to protect the lanthanide from quenching from vibrational energy dissipation by oscillators like O-H of water. The antenna harvests energy through high molar absorption to the singlet excited state. After undergoing intersystem crossing to the triplet state, the antenna transfers energy to the excited state 5DJ state of the lanthanide. The radiative transition of electrons from the excited 5DJ state to the 7FJ states results in luminescent emission from the lanthanide ion. (B) Luminescent 4f-4f transitions of europium and terbium complexes and commonly observed emission wavelengths to emit red and green light, respectively.,

Pendant antennae used to sensitize lanthanide complexes. (A) Schematic of lanthanide chelates that utilize pendant antennae. The pendant antenna is attached to the chelate (e.g. DO3A or DTPA) with a linker (top), and multiple pendant antennae can be incorporated (bottom)., (B) Examples of common sensitizers used as pendant antennae. Pendant antennae are derived from organic fluorophores based on favorable photophysical properties for sensitization.

Bright Eu(III) luminescence probes that incorporate multiple pendant antennae into a triazacyclononane chelate. Alkynylpyridyl units are used as antennae that coordinate to the Eu(III) center through the pyridine nitrogen to decrease metal-antenna distance and increase electron transfer efficiency (ηET). Further, Eu(III)-coordinating carboxylate or phosphinate units are p-substituted on the pyridine moiety to achieve coordinative saturation and prevent solvent quenching.

Chelating antennae used to sensitize lanthanide complexes. (A) Schematic of a chelating antennae that serves dual function as both the protective lanthanide chelate and the sensitizing antenna. The decreased metal-antenna distance affords efficient energy transfer. (B) Examples of recently reported lanthanide complexes with chelating antennae. The chelating antennae based on the IAM and 1,2-HOPO binding units (top, in green and red, respectively) coordinate to the metal centers through the through salicylate- and catecholate-type coordination, respectively.,, The poly-acid based chelates (bottom) by Yuan and coworkers coordinate to the lanthanide center through the pyridine/pyrazole moieties and the carboxylates.

Chiral IAM tetradentate chelates with strong CPL signal. The CPL signal is highly influenced by the rigidity of the ethylenediamine backbone (black). The oxygens labeled in blue coordinate to the metal center in a salicylate-type manner.

Sensitization bias of varying carbostyril derivatives. Tremblay et al. demonstrated the principle of ratiometric detection by antenna modification. Altered functionalities on the antenna exhibit differential sensitization of Tb(III) and Eu(III) luminescence. This can be harnessed for ratiometric switches. (Reprinted with permission from reference . Copyright 2007 American Chemical Society.)

General scheme for luminescent lanthanides that respond to an analyte by modulation of q, the number of waters bound. Water can quench luminescence through promoting energy dissipation by vibrational transfer to the O-H oscillator. Replacement of water with an anionic analyte of interest can increase emission intensity through relieving quenching by this process. Furthermore, ligand exchange of water leads to change in ligand polarization and thus, emission bands corresponding to electric dipole transitions.

A pH-responsive lanthanide luminescent probe that localizes in the lysosomes. The amide-linked azaxanthone sensitizer promotes cell uptake through macropinocytosis, resulting in trafficking through the mitochondria and finishing in the lysosomes. The sulfonamide moiety confers pH-sensitivity to the probe: in acidic conditions, the sulfonamide is protonated, precluding coordination to the lanthanide ion and opening a coordination site for one water molecule. As the probe encounters a higher pH environment, the sulfonamide is deprotonated, permitting coordination through the nitrogen and displacement of the bound water.

A bright pH-responsive europium luminescent probe that localizes in the endoplasmic reticulum. High quantum yields were achieved by incorporating mutiple aryl-alkynyl groups as sensitizers (quantum yield = 50% compared to the azaxanthone at 6% or less). In combination with the pyridyl groups, the aryl-alkynyl groups localize the probes in the endoplasmic reticulum. The sulfonamide moiety confers pH-sensitivity to the probe similar to the azaxanthone-containing pH probe.

Examples of oxyanions that have been detected with luminescent lanthanide probes through q-modulation. Sterics and charge of the lanthanide complex govern selectivity for the target anion.

Comparison of binding affinities of citrate and lactate for a series of oxy-anion binding Eu(III) complexes. Ratios of citrate to lactate binding affinities are calculated from log Ka reported in reference and represented in terms of ligand modifications and overall charge.

Bicarbonate-selective complexes that localize in the mitochondria. Bicarbonate serves as an anionic analyte that can displace the bound water molecules and change spectra form and emission intensity. The amide-linked azaxanthone encourages macropinocytosis and consequential localization in the mitochondria. Steady-state concentrations of bicarbonate could be ratiometrically measured ΔJ=2/ΔJ=1 emission intensities of the Eu(III) complex.

Luminescent lanthanides that respond to an analyte by decreasing the distance between the metal center and the antenna. In the absence of the analyte, the antenna is either not attached to the lanthanide chelate or separated by a flexible linker, resulting in low efficiency in energy transfer (and sensitization). The analyte brings the antenna in close proximity to the lanthanide, increasing the efficiency of energy transfer and emission intensity.

Structure and mode of action of the distance-deEu-KPhen. In the presence of potassium, concomitant complexation of the ion by the crown ether and the phenanthridine (through a cation-π interaction) decreases the distance between the antenna and the Eu(III) center, increasing luminescence intensity.

Copper(I)-sensitive luminescent lanthanide that responds by decreasing the distance between the antenna and the metal center. A drastic “switch-on” response is observed through a copper-dependent “click” reaction that attaches the dansyl azide antenna to the Eu(III) chelate and permits energy transfer.

Modulation of antenna-Ln(III) distance by reversible coordination of the antenna. (A) “Switch on” designs involve the coordination of a sensitizing analyte such as aromatic carboxylates whereas (B) “switch off” probes rely on the displacement of the antenna by an anionic analyte.

Modified reaction-based approach for the detection of non-sensitizing analytes by reversible antenna coordination. (A) Meyer and Karst detected peroxidase activity using a pro-antenna, p-hydroxyphenylpropionic acid as a substrate. In the presence of the enzyme, the pro-antenna dimerizes to the biphenol that can coordinate and sensitize Tb(III) EDTA. (B) Page et al. detected hydroxyl radicals through reaction with the trimesate pro-antenna to form the 2-hydroxytrimesic acid antenna that can coordinate to Tb(III) DO3A.

Gunnlaugsson and coworkers have used coordinating antennae to detect dicationic transition metals [M(II)]., Selectivity is influenced by affinity of the ligand for the transition metal ion. In the absence of M(II), the antenna coordinates the Eu(III) center and sensitizes luminescence. Preferred M(II) can compete for coordination of the antenna to switch off the Eu(III) luminescence, and in some cases, produce a luminescent d-metal complex.

(A) Bioresponsive lanthanides that employ caged antennae., In the absence of an analyte, the antenna precursor is not an efficient sensitizer. The analyte reacts with the cage to trigger formation of a sensitizing chromophore, producing Ln(III) luminescence. (B) The caged antenna approach for the detection of hydrogen peroxide. The boronate ester mask is chemoselective for hydrogen peroxide. Analyte reaction results in an aniline or phenol that can efficiently sensitize Tb(III) luminescence.

Antenna-triggered formation of coumarin sensitizer for the detection of a series of analytes. Synthetic procedures were simplified by using an azide-modified chelate that could be attached to an alkyne-modified antenna building block. The antenna building block (blue) was derivatized with either phenylboronic acid, triisopropylsilane, allyl, or galactose cages for the detection of hydrogen peroxide, fluoride, palladium, or β-galactosidase, respectively. In the presence of the analyte, the cage is removed driving formation of the coumarin antenna (orange) for sensitized Ln(III) luminescence.

PeT-switchable luminescent lanthanides., In the “switched-off” state of a PeT-switchable emissive lanthanide system, the HOMO of the PeT switch is higher in energy than the S0 ground state of the antenna (left). The PeT switch donates an electron to pair to the S0 ground state of the antenna, quenching its fluorescence. The PeT process competes with radiative decay and reduces the sensitizing efficiency of the antenna and, consequently, lanthanide luminescence. When an analyte chemically alters the PeT switch to decrease its HOMO energy, it can no longer serve as an electron donor to the antenna (right). In turn, the antenna’s sensitizing capacity is restored and the lanthanide luminescence is “switched on”.

Investigating the PeT switch mechanism for modulating Ln(III) luminescence. In a systematic investigation, Terai et al. evaluated the PeT quenching of a quinolinone antenna by a series of organic moieties with varying HOMO energies. The PeT switch approach was validated in this study, as decreasing HOMO energies (which is expected to correspond to decreased donor capacity and decreased quinolone quenching) increased the observed luminescence. (Adapted with permission from reference . Copyright 2006 American Chemical Society.)

PeT switchable Ln(III) complexes with chelating polyacid antennae. In the general design, a PeT switch is conjugated to the central pyridine of the chelate.,,,– Analyte interaction with the PeT decreases electron transfer to the antenna and increases luminescence intensity. (A) The chelated complex is cell-impermeable so a method of administration was devised wherein the cells are simultaneously treated with the free Ln(III) salt and an ester-protected form of the chelate. Upon cell internalization, intracellular esterases cleave the ester protecting groups to expose the polyacids, permitting self-assembling of the chelate in the cell. This strategy has been used for the detection of several reactive small molecules, such as (B) HOCl and (C) NO.

ATP-selective probe driven by PeT from the adenosine of ATP to the phenanthridine antenna. Nucleoside phosphate selectivity over other anionic phosphates is partially driven by π-π stacking interactions between the adenosine and the phenanthridine antenna. Further, this antenna-nucleoside interaction alters luminescence by PeT from the analyte to the antenna (decreasing luminescence). The tricationic charge of the Tb(III) complex confers selectivity for the 4- charge of ATP (over the 3- and 2- charge of ADP and AMP, respectively).

Modulation of antenna sensitization with an analyte receptor unit for the detection of Cu(II) or Hg(II). The iminodiacetate receptor unit can coordinate to d-metal ions. Coordination of Cu(II) or Hg(II) alters the excited state of the phenyl antenna and quenches Tb(III) luminescence.

Schematic for analyte detection by CPL “fingerprints.” Binding of a chiral analyte (through either direct metal coordination or non-covalent interactions) to a luminescent lanthanide can introduce detectable chirality to or alter the chiroptical properties of the lanthanide complex. As a result, the analyte-bound complex produces detectable circularly polarized luminescence.

Chiral Eu(III) probe for the detection of “acute-phase” proteins by CPL. Protein binding occurs through hydrophobic interactions with the azaxanthone or azathioxanthone antenna units and coordinative interactions between the Eu(III) center and a glutamate side chain. The protein alters the chirality of the chelate, inducing a change in CPL signal profile and intensity.

CPL-active Eu(III) probes for protein labeling. The approach utilizes the CPL fingerprints of different proteins to identify the analytes. The antenna is first labeled with the protein of interest through a peptide bond. The addition of the antenna to a Eu(III) chelate leads to self-assembly of the luminescent lanthanide with well-defined CPL signal.

When placed in a magnetic field I = ½ nuclei will either align with or against the field. The population of the two states is determined by the Boltzmann distribution; consequently, slightly more nuclei will occupy the lower energy state, i.e., the state aligned with the magnetic field.

A.) The spin angular momentum of each proton nucleus creates a magnetic moment (µ) which, in the presence of an external magnetic field (B0), precesses about the z-axis with a characteristic frequency (ω0). B.) In a macroscopic sample, the magnetic moment of the individual nuclei can be summed into a single magnetization vector which lies along the z-axis. C.) The application of an RF pulse creates a secondary magnetic field (B1) which rotates the magnetization vector away from the z-axis, tipping it toward the xy plane. D.) Assuming the application of a 90° pulse, immediately after the RF field is removed, the magnetization vector will lie in the xy plane. E.) Over time, various relaxation processes return Mx, My, and Mz to their equilibrium values. Spin-lattice, or T1, relaxation processes are responsible for the recovery of the Mz component of the magnetization vector. Spin-spin, or T2, relaxation processes are responsible for returning Mx and My to zero.

Immediately following the removal of the RF field, there is an equal distribution of spins in the high and low energy state, resulting in a net Mz of 0. A number of nuclei must transition back to the low energy state to restore the initial Boltzmann distribution and return Mz to its equilibrium value. These transitions occur when the nuclei undergo stimulated emission, mediated by surrounding magnetic fields fluctuating at the Larmor frequency (ω0).

Gd(III)-DOTA is a clinically approved contrast agent where the Gd(III) ion is chelated by a macrocyclic polyaminocarboxylate ligand, leaving a single coordination site open on the lanthanide for water access.

There are three parameters that influence r1 that can be modified by structural changes to the chelate structure: q, the number of water molecules directly bound to the Gd(III) center; τm, the inverse of the rate of exchange of the bound water with the bulk solvent; and τR, the rotational correlation time of the complex, or how rapidly it tumbles in solution.

The biphenylcyclohexyl moiety of MS-325 binds to HSA, a 67 kDa protein.,

Bz-CB-TTDA, an HSA-binding derivative of TTDA.

The n-CHTGd series of probes forms a covalent bond with the reporter protein HaloTag, undergoing a 6-fold increase in relaxivity upon protein binding.

Strategies for selective conjugation of the HOPO ligand to the interior or exterior surface of MS2 viral capsids. (Figure reproduced with permission from reference . Copyright 2008 American Chemical Society.)

Alkyne modified Gd-DO3A chelates were conjugated to azide-functionalized DNA strands using copper-catalyzed click chemistry. The Gd(III)-modified DNA was then immobilized on 30 or 13 nm AuNPs. All relaxivity values are in mM−1s−1, were acquired at 60 MHz and are reported per Gd(III) ion.

By peptide coupling Gd(III) chelates to quadruplex-forming oligonucleotides, Hamilton et al. developed macromolecular structures with high relaxivity, biocompatibility and modularity.

Lipophillic complexes developed by Botta et al. The introduction of two aliphatic chains restricts rotation of the chelate once it has been incorporated into a liposome or micelle.

Multimeric Gd(III) complexes conjugated to organic scaffolds via copper-catalyzed click chemistry. The relaxivity values were measured at 60 MHz and are reported per Gd(III) ion.–

Metallostar structures that have been developed and the parameters of interest. All relaxivity values were measured at 20 MHz unless otherwise noted, and are given on a per Gd(III) basis. A and B utilize a DTTA chelate with a q = 2,, leading to high r1 values; however, by changing the ligand from a terpyridine (tpy) to bpy and extending the linker between the Gd(III) chelate and the ligand, Tóth and Ruloff were able to increase the log KDGdL from 10.87 to 18.2.–

The tetraamide derivative of Gd(III)-DOTA displays dramatically slower water exchange than the anionic Gd(III)-DOTA.

Sherry et al. computed the Mulliken charge of the chelating oxygen atom for Eu(III) chelates with pendant carboxylate, amide and ketone arms. The carboxylate oxygen had the largest partial negative charge, the ketone oxygen (or the enol tautomer) had the smallest negative charge, and the amide oxygen was intermediate between the two. A systematic investigation of a series of chelates with pendant amide and ketone arms confirmed that incorporation of less negatively charged ketone groups results in slower water exchange, likely due to a stronger Gd-Ow resulting from a more electropositive metal center.

Schematic of library of peptoid-modified Eu(III) DOTA – tetraamide derivatives.

Sterically crowded structures studied by Merbach et al. that exhibit optimal water exchange rates. Incorporation of an additional methylene group (shown in red).–

The SAP (M) and TSAP (m) isomers of macrocyclic lanthanide chelates differ in the twist angle between the planes defined by the chelating nitrogen and oxygen atoms. It is proposed that the TSAP isomer is more sterically crowded about the water binding site, weakening the Gd-Ow bond and leading to faster water exchange.

SAP/TSAP interconversion occurs by either arm rotation or ring inversion. Sequential application of both processes leads to enantiomerization. (Figure adapted with permission from reference . Copyright 2012 American Chemical Society.)
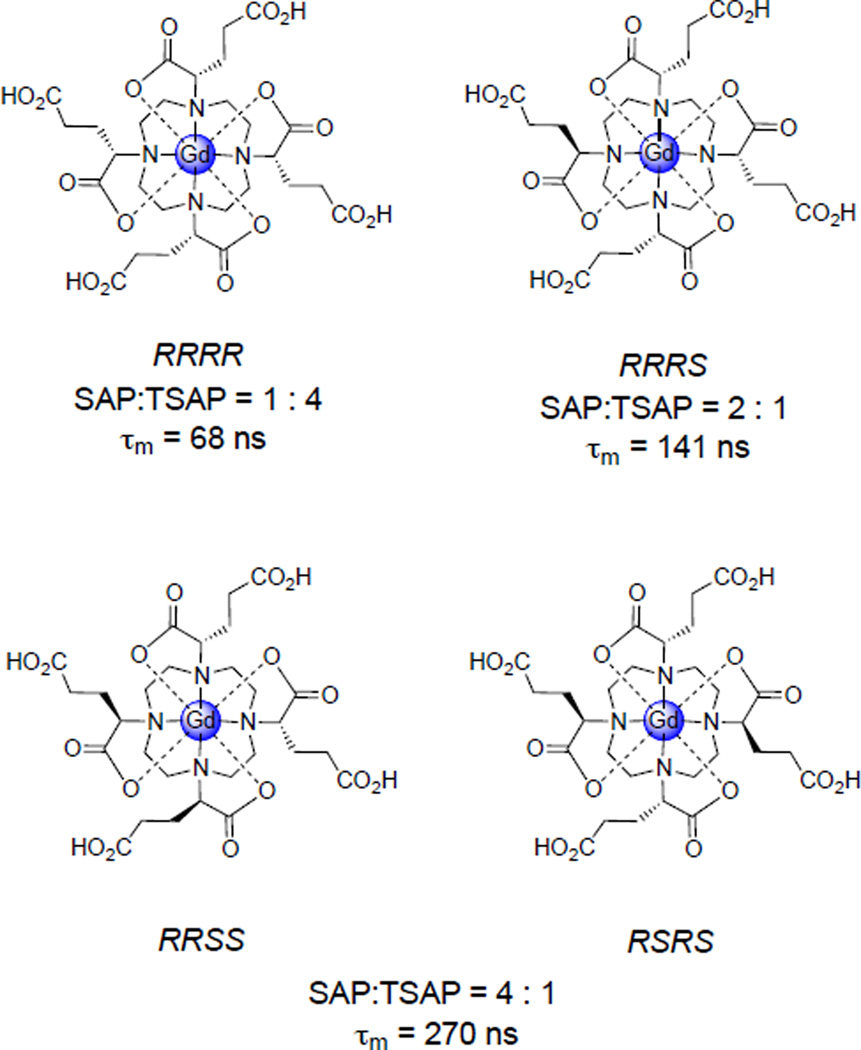
Woods et al. examined the SAP : TSAP ratio and water exchange kinetics of three derivatives of Gd(III)-DOTA that incorporate a carboxyethyl substituent α to the macrocycle and determined that the complexes with lower SAP : TSAP ratios exhibit significantly faster water exchange.

Substitution of carboxylate groups for two of the amide back-binding groups of DOTAM magnifies the difference in water exchange rate of the SAP and TSAP isomers.

(A) The process of ring inversion exchanges axial protons (Ha and Hd, shown in red) with equatorial protons (Hc and Hb, shown in blue) and vice versa. (Figure adapted with permission from reference . Copyright 2000 American Chemical Society.) (B) During the ring inversion process, the axial and equatorial protons of the two isomers exchange with one another; conversely, during the arm rotation process, the axial protons of one isomer exchange only with the axial protons of the other. (Figure reproduced with permission from reference . Copyright 2010 American Chemical Society.)

Substituted DOTA derivatives that restrict isomerization processes. Substitution on the backbone of the macrocycle (shown in orange) restricts ring inversion, while substitution of the pendant carboxylates/amides of the chelate (shown in blue) prevents arm rotation. Substitution at both positions effectively locks the chelate in a single conformation, either SAP or TSAP, depending on the absolute stereochemistry at the substituted positions. S-SSSS p-NO2-Bn-DOTMA (boxed in red) is effectively locked in the desired TSAP conformation.

(A) τr-modulated contrast agents undergo an increase in molecular weight upon activation by the event of interest, causing an increase in τr and consequently relaxivity. Increase in MW typically occurs through (B) protein binding or (C) self-assembly due to a biological event of interest.

In response to the biological event of interest, q-modulated contrast agents transform from a low-relaxivity q = 0 state to a high relaxivity q = 1 state.,, (Figure reproduced with permission from reference . Copyright 2013 American Chemical Society.)

Example of a system in which CEST can occur. Water protons and amine protons resonate at different NMR frequencies and can undergo chemical exchange with one another. If the water protons (HB) are selectively saturated, chemical exchange will transfer some of the magnetization saturation to the amine protons (HA), allowing for indirect saturation of the signal from HA.,

A presaturation pulse applied to Pool B increases the number of spins aligned against the magnetic field in this pool (middle). The distribution of the spins in Pool A will depend upon the longitudinal relaxation rates of both Pool A and Pool B. If the relaxation rate of either pool is faster than the rate constant of exchange, then the system will relax back to the Boltzmann distribution before exchange can transfer the altered spin state to Pool A (top). The result will be a normal NMR spectrum. If relaxation is slow compared to chemical exchange, then the high energy level spins will equilibrate thus reducing the bulk magnetization in this pool (right). The result will be signal intensity reduction in both Pool A and Pool B.

CEST spectrum of a 30 mM solution of a tetraamide Eu(III) analogue, Eu(III)-DOTA-4AmCE3+, acquired at 270 MHz and 25 °C. The sharp signal reduction at 0 ppm is due to direct saturation of the bulk water signal, whereas the sharp reduction near +50 ppm is due to the PARACEST effect arising from saturation of water molecule coordinated to the lanthanide that is in exchange with the bulk solution., Image adapted from reference with permission. Copyright 2006 Royal Society of Chemistry).

Spectrum showing change in CEST before (black) and after (red) covalent binding of Tm-DO3A-cadaverine (right) to glutamine side chains of albumin by transglutaminase. Upon creation of the new bond by the enzyme, CEST decreased at +4.6 ppm and increased at −9.2 ppm., (Figure reproduced with permission from reference . Copyright 2013 American Chemical Society.)

The Ca(II)-sensitive DOPTA-Gd, a q-modulated probe, exhibits a 75% increase in relaxivity in the presence of physiologically relevant concentrations of Ca(II).,,

The relaxivity of the Zn(II) activated Gd-DOTA-diBPEN, a τr modulated contrast agent, increases more than 150% upon the addition of Zn(II), in the presence of HSA.

In an ex vivo experiment, Gd-DOTA-diBPEN was able to detect changes in extracellular Zn(II) concentration following the addition of an insulin-stimulating concentration of glucose to isolated β-islet cells. (Figure reproduced with permission from reference . Copyright 2012 Elsevier.)
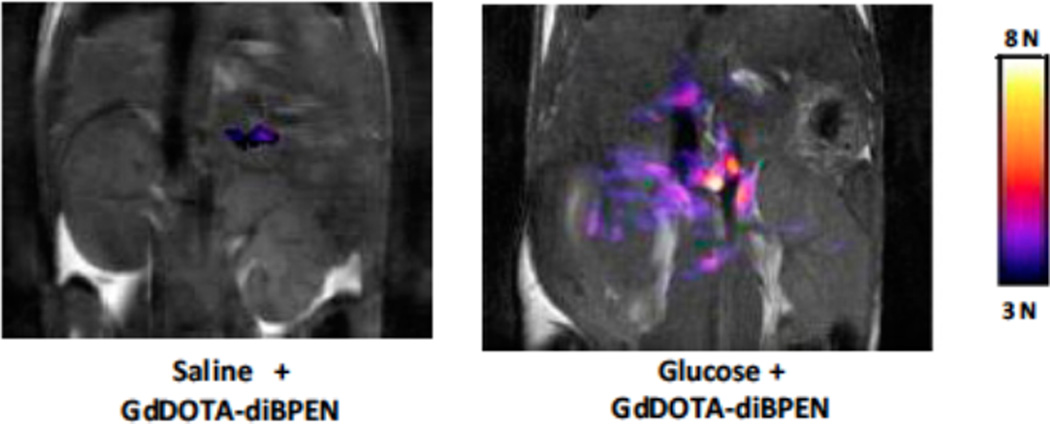
In vivo images acquired by Sherry et al. of the abdominal region of a 12-week old mouse following IP injection of either saline (left) or glucose (right) and subsequent tail vein injection of the Zn(II)-activated Gd-DOTA-diBPEN. Significant contrast enhancement in the area of the pancreas was observed following the administration of glucose and Gd-DOTA-diBPEN, indicating that the agent is capable of detecting fluctuations in the concentration of Zn(II) associated with insulin release by β-islet cells. (Figure reproduced with permission from reference . Copyright 2012 Elsevier.)

The Gd-daa-n series of Zn(II) activated contrast agents developed by Meade et al. consists of a Zn(II) binding domain conjugated to a Gd-DO3A chelate by an aliphatic linker (top). The change in relaxivity upon the addition of Zn(II) strongly depends on the length of the aliphatic linker, n. Figure reproduced with permission from reference . Copyright 2013 American Chemical Society.

Copper-activated MR contrast agents developed by Chang et al., which show remarkable affinity and selectivity for copper over other biologically relevant metal cations as well as an extremely large change in relaxivity in response to the presence of copper.,,

PARACEST spectrum of 5 mM Yb(S-THP)3+ at pH 7.0 before (closed circles) and after (open circles) the addition of 10 mM diethyl phosphate (DEP). The PARACEST peak arising from the exchangeable alcohol protons (near 20 ppm) appears only after the addition of DEP.– (Figure reproduced with permission from reference . Copyright 2010 American Chemical Society.)

The EGad series of bioresponsive MRI contrast agents responds to the presence of β-galactosidase.– The first generation, EGad (left), showed only a 20% increase in relaxivity in the presence of β-gal. The introduction of a methyl group to the linker between the chelate and the sugar moiety, either α (center) or β (right) to the macrocycle, increases this Δr1 to 40–50%.

In the presence of β-glucoronidase, the sugar moiety of the agent depicted is cleaved, initiating an electron cascade that causes the dissociation of the self-immolative linker.,

In the presence of β-galactosidase, the sugar moiety of Gd(DOTA-FPG) is cleaved. Subsequent rearrangement results in the formation of a highly reactive electrophile that is rapidly attacked by a nucleophilic site on HSA (or another available protein) to form a high molecular weight adduct.

Following enzymatic cleavage of the sugar substrate, the complex terminates in a hydrophobic biphenyl moiety that has a high affinity for HSA. Once the complex forms a non-covalent adduct with the protein, both the τR and the relaxivity of the complex increase.

In the presence of MPO, the 5-HT moieties of Gd-MPO become oxidized and form radicals. Subsequent radical polymerization produces oligomers up to five units long. These oligomers can then become cross-linked through interactions with endogenous proteins, further increasing the molecular weight, and consequently the τR and relaxivity, of the contrast agent.–

τR-modulated, pH responsive probe developed by Aime et al.,, As the pH increases, the peptide structure becomes more rigid, increasing both the τR and the relaxivity of the complex.

pH responsive PARACEST probe, Ln(III)-DOTAM-Gly, containing two pools of exchangeable protons: those of the coordinated water molecule (red) and those of the amide groups on the pendant arms (blue). These give rise to two separate PARACEST signals, the intensity of which can be monitored independently to give a ratiometric measure of pH.–

As the pH increases, the phenol (red) becomes deprotonated (pKa ~6.5–6.7), allowing the complex to adopt a quinone like resonance structure in which the chelating ketone becomes negatively charged (blue), dramatically altering the coordination environment about the lanthanide center. This in turn changes both the frequency and the intensity of the PARACEST signal from the coordinated water molecule, allowing for ratiometric determination of pH from interrogation of a single pool of protons whose PARACEST signal is significantly shifted from the bulk water resonance.
References
-
- Bünzli J-CG, Eliseeva SV. In: Springer Series on Fluorescence. Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects. Hänninen P, Härmä H, editors. Vol. 7. Springer Verlag: Verlag Berlin Heidelberg; 2011. pp. 1–46.
-
- Ramogida CF, Orvig C. Chem. Commun. 2013;49:4720. - PubMed
-
- Laurent S, Vander Elst L, Muller RN. Q. J. Nucl. Med. Mol. Imaging. 2009;53:586. - PubMed
-
- Hanaoka K. Chem. Pharm. Bull. 2010;58:1283. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
