

Combined approaches to flexible fitting and assessment in virus capsids undergoing conformational change
- PMID: 24333899
- PMCID: PMC3988922
- DOI: 10.1016/j.jsb.2013.12.003
Combined approaches to flexible fitting and assessment in virus capsids undergoing conformational change
Abstract
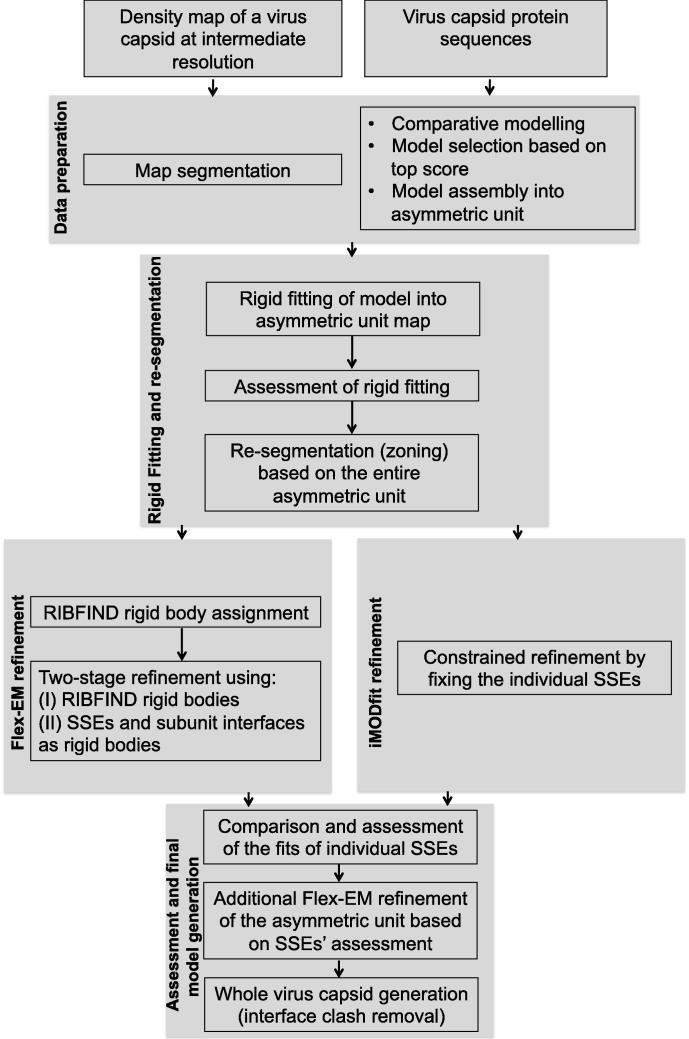
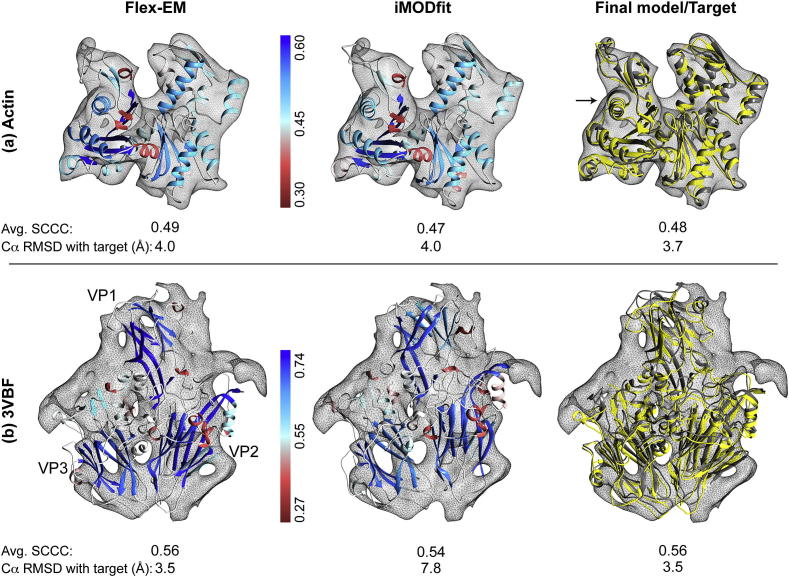
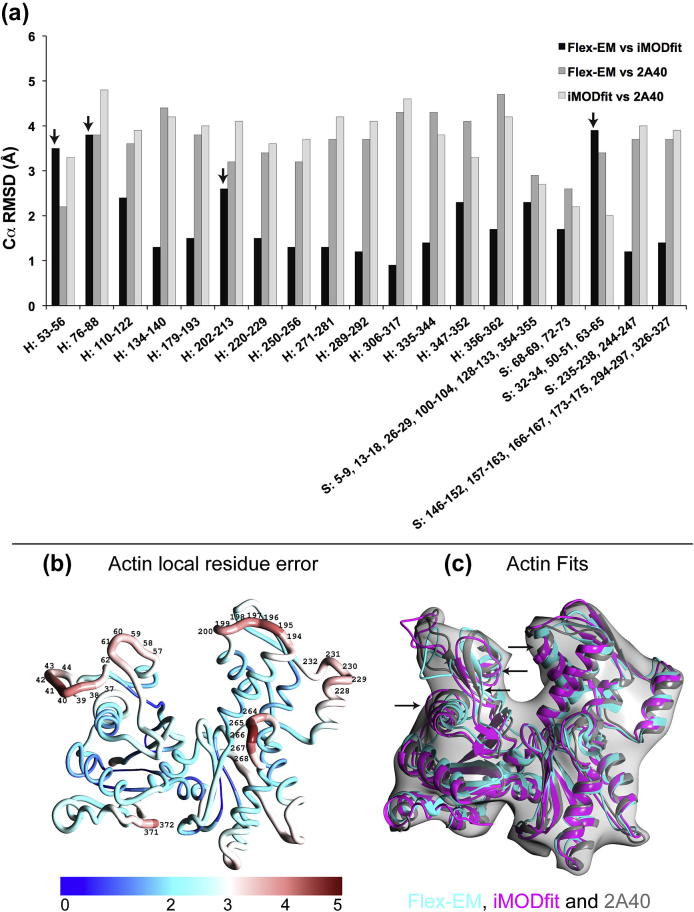
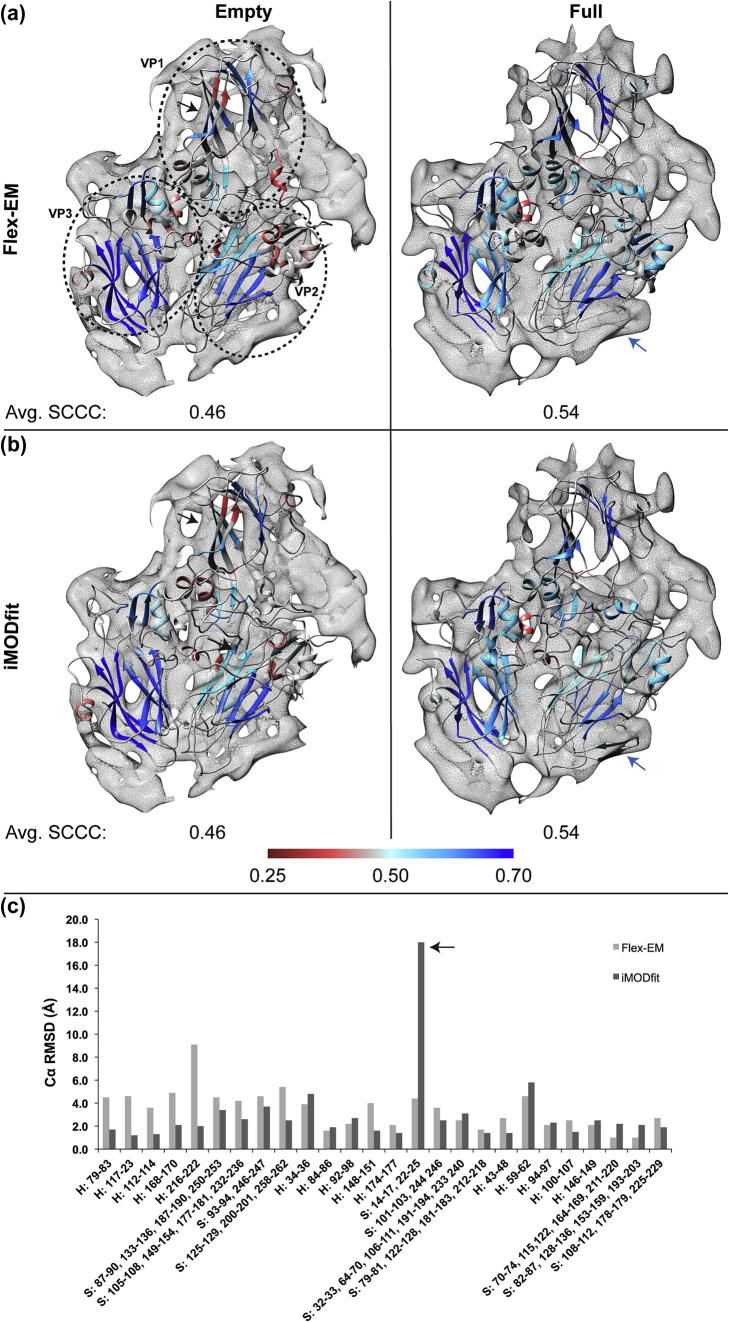
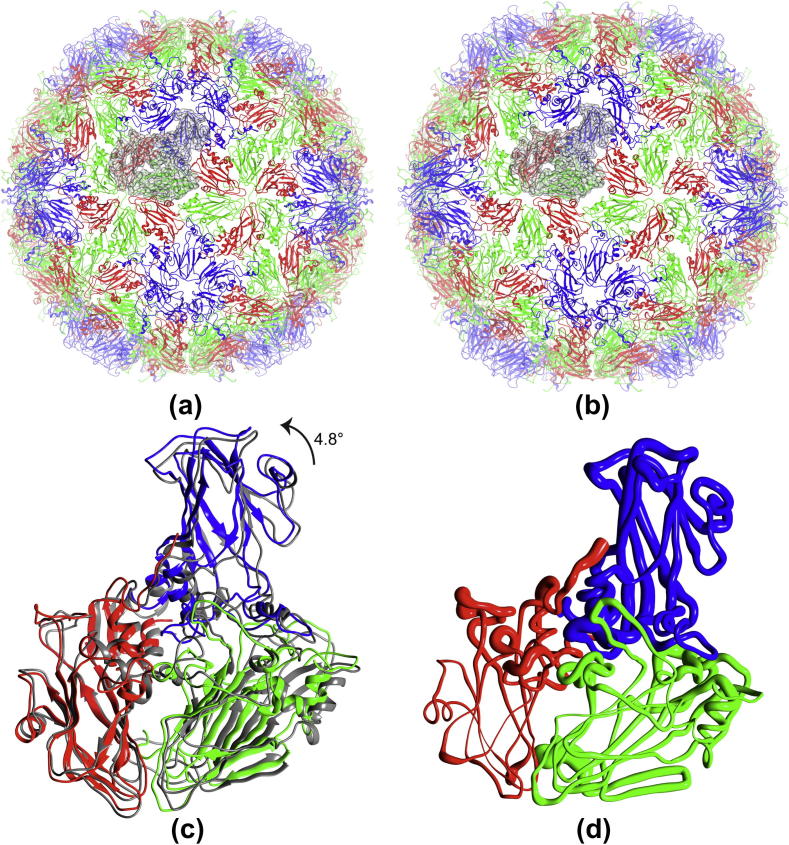
Fitting of atomic components into electron cryo-microscopy (cryoEM) density maps is routinely used to understand the structure and function of macromolecular machines. Many fitting methods have been developed, but a standard protocol for successful fitting and assessment of fitted models has yet to be agreed upon among the experts in the field. Here, we created and tested a protocol that highlights important issues related to homology modelling, density map segmentation, rigid and flexible fitting, as well as the assessment of fits. As part of it, we use two different flexible fitting methods (Flex-EM and iMODfit) and demonstrate how combining the analysis of multiple fits and model assessment could result in an improved model. The protocol is applied to the case of the mature and empty capsids of Coxsackievirus A7 (CAV7) by flexibly fitting homology models into the corresponding cryoEM density maps at 8.2 and 6.1Å resolution. As a result, and due to the improved homology models (derived from recently solved crystal structures of a close homolog - EV71 capsid - in mature and empty forms), the final models present an improvement over previously published models. In close agreement with the capsid expansion observed in the EV71 structures, the new CAV7 models reveal that the expansion is accompanied by ∼5° counterclockwise rotation of the asymmetric unit, predominantly contributed by the capsid protein VP1. The protocol could be applied not only to viral capsids but also to many other complexes characterised by a combination of atomic structure modelling and cryoEM density fitting.
Keywords: Coxsackievirus A7; Electron cryo-microscopy; Flexible fitting; Model assessment; Picornaviridae.
Copyright © 2013 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Abrescia N.G., Cockburn J.J., Grimes J.M., Sutton G.C., Diprose J.M. Insights into assembly from structural analysis of bacteriophage PRD1. Nature. 2004;432:68–74. - PubMed
-
- Ahmed A., Tama F. Consensus among multiple approaches as a reliability measure for flexible fitting into cryo-EM data. J. Struct. Biol. 2013;182:66–67. - PubMed
-
- Bajaj C., Yu Z., Auer M. Volumetric feature extraction and visualization of tomographic molecular imaging. J. Struct. Biol. 2003;144:132–143. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
