

TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy
- PMID: 24335147
- PMCID: PMC3950670
- DOI: 10.1093/nar/gkt1260
TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy
Abstract
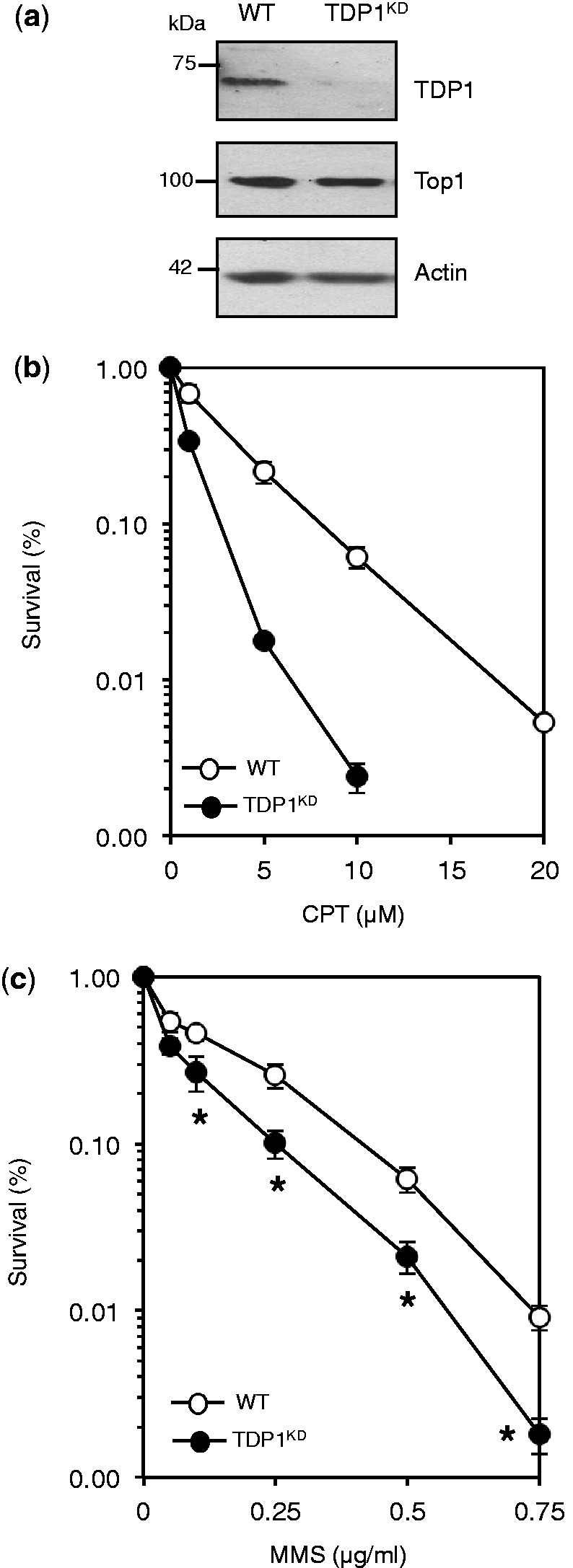
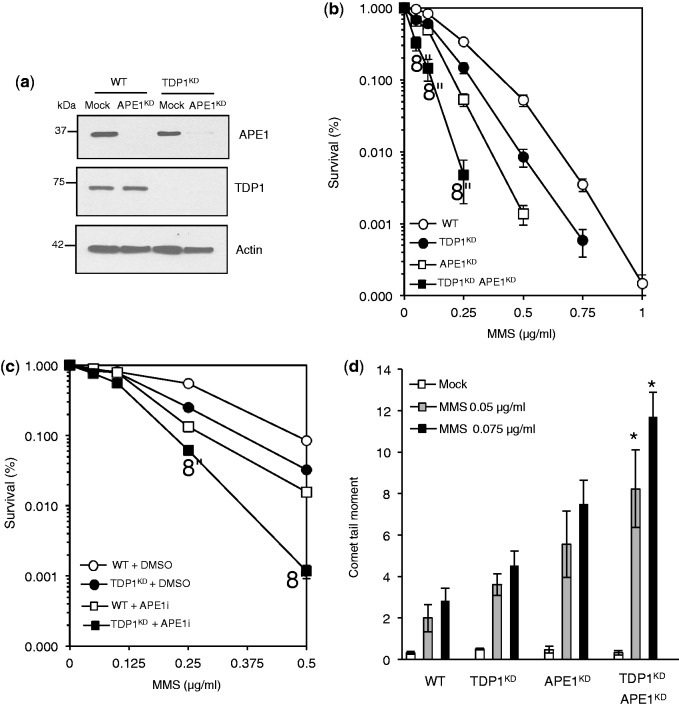
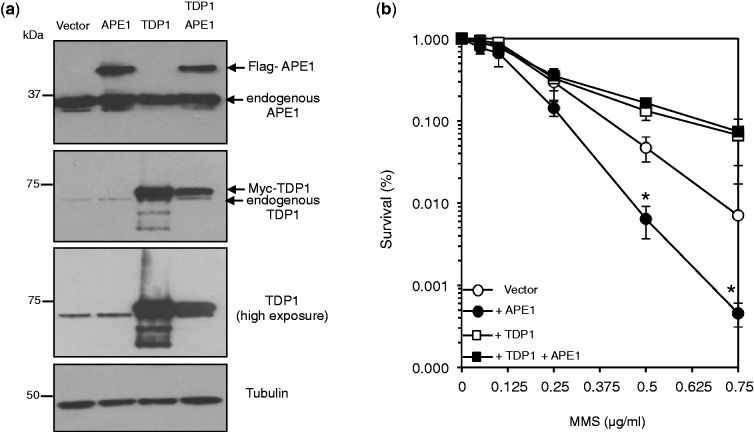
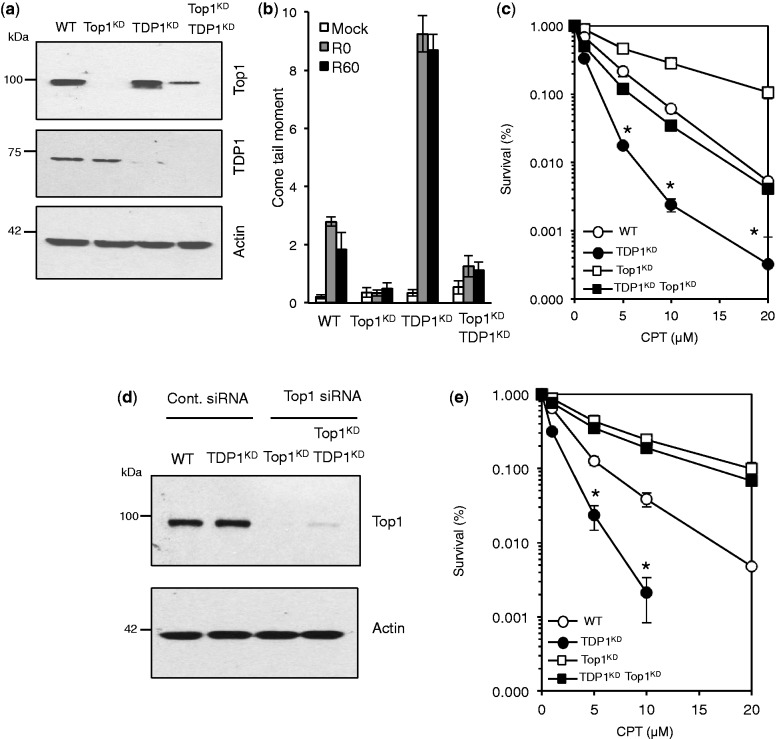
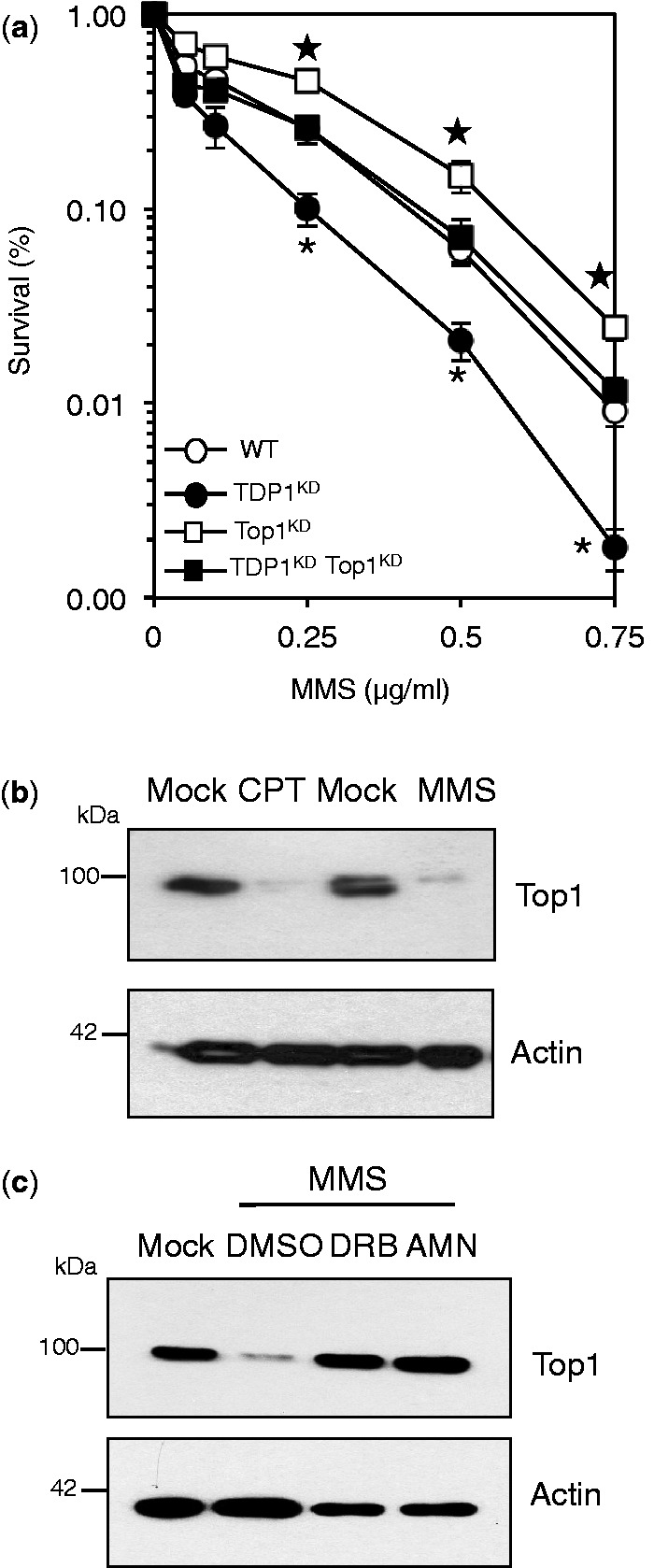
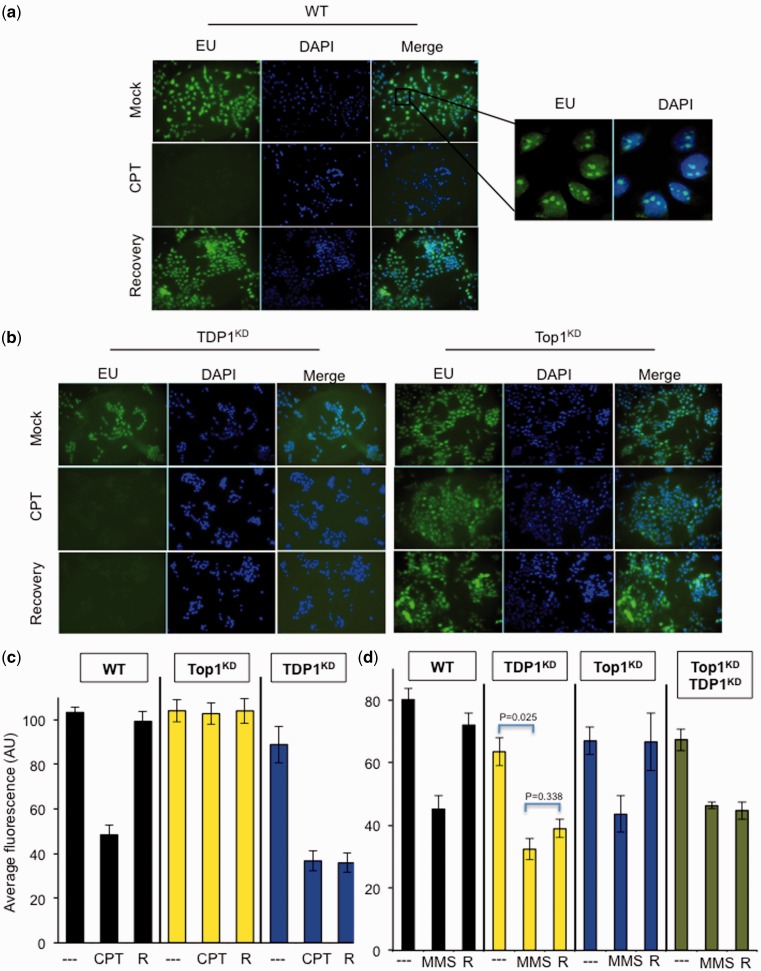
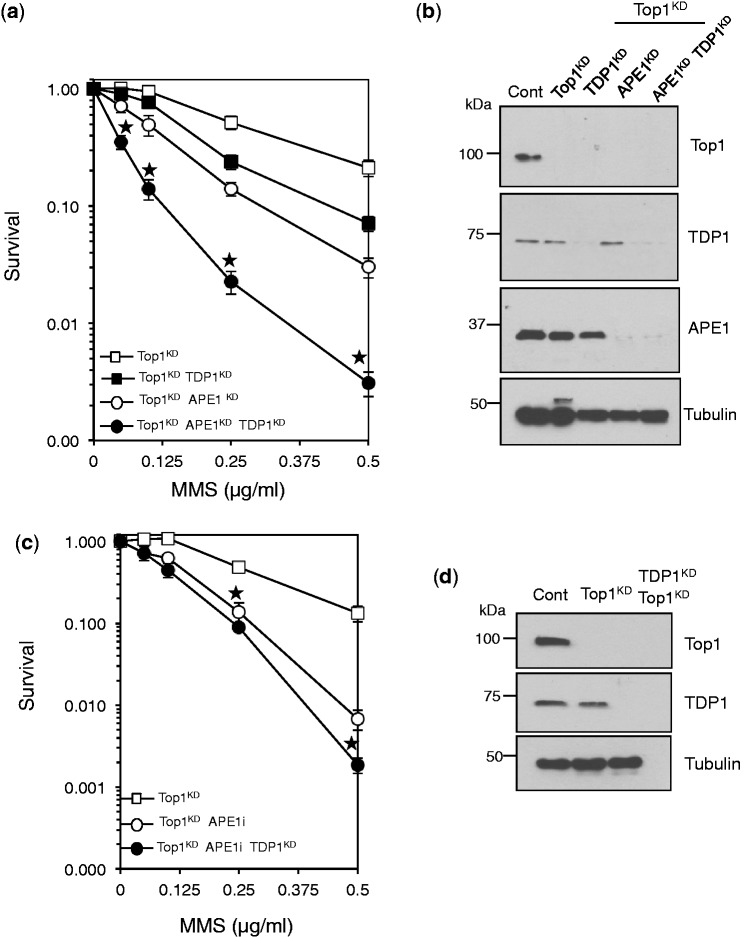
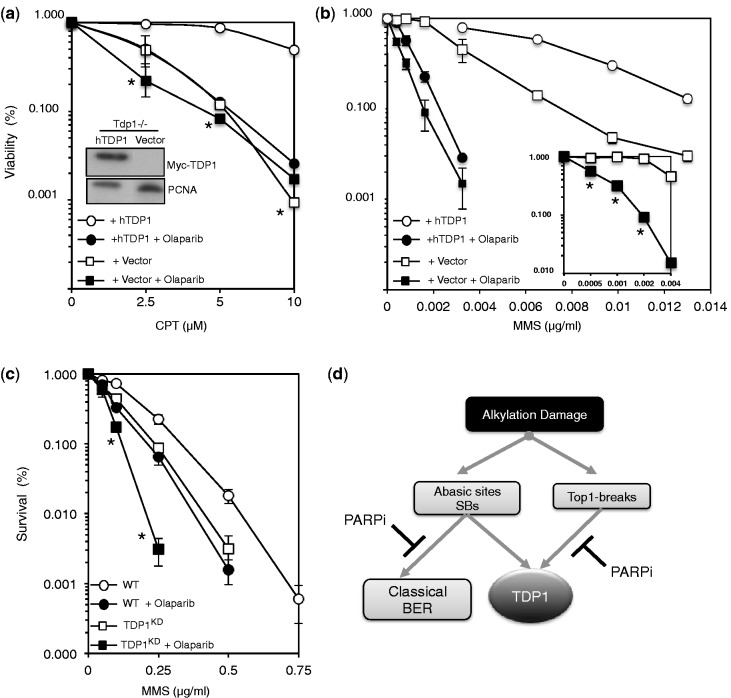
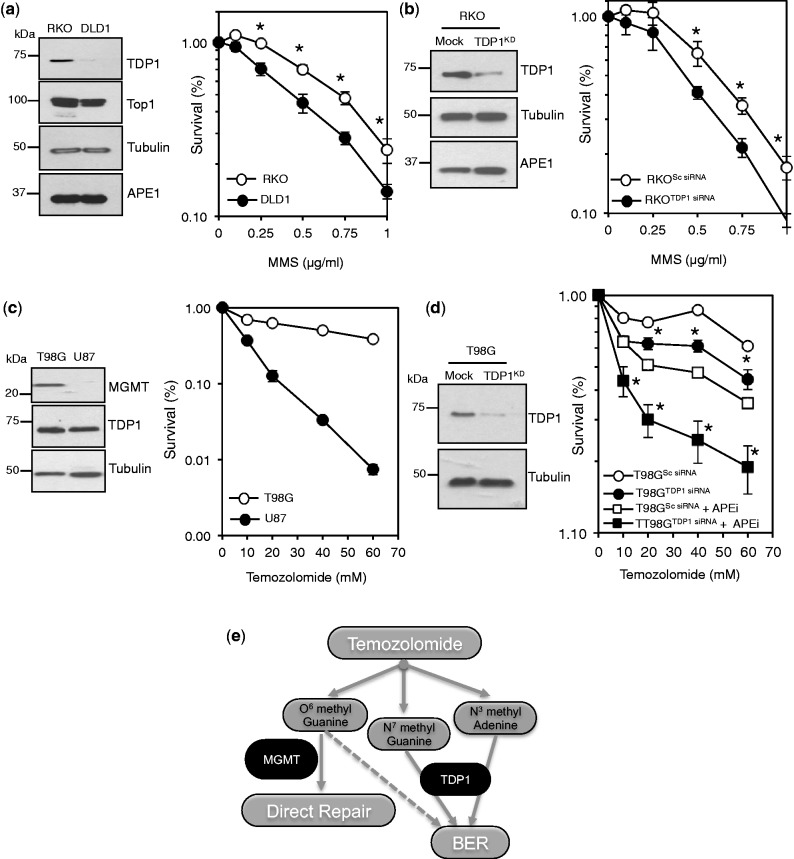
Base damage and topoisomerase I (Top1)-linked DNA breaks are abundant forms of endogenous DNA breakage, contributing to hereditary ataxia and underlying the cytotoxicity of a wide range of anti-cancer agents. Despite their frequency, the overlapping mechanisms that repair these forms of DNA breakage are largely unknown. Here, we report that depletion of Tyrosyl DNA phosphodiesterase 1 (TDP1) sensitizes human cells to alkylation damage and the additional depletion of apurinic/apyrimidinic endonuclease I (APE1) confers hypersensitivity above that observed for TDP1 or APE1 depletion alone. Quantification of DNA breaks and clonogenic survival assays confirm a role for TDP1 in response to base damage, independently of APE1. The hypersensitivity to alkylation damage is partly restored by depletion of Top1, illustrating that alkylating agents can trigger cytotoxic Top1-breaks. Although inhibition of PARP activity does not sensitize TDP1-deficient cells to Top1 poisons, it confers increased sensitivity to alkylation damage, highlighting partially overlapping roles for PARP and TDP1 in response to genotoxic challenge. Finally, we demonstrate that cancer cells in which TDP1 is inherently deficient are hypersensitive to alkylation damage and that TDP1 depletion sensitizes glioblastoma-resistant cancer cells to the alkylating agent temozolomide.
Figures
References
-
- Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. - PubMed
-
- Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999;424:127–142. - PubMed
-
- Kato S, Bowman ED, Harrington AM, Blomeke B, Shields PG. Human lung carcinogen-DNA adduct levels mediated by genetic polymorphisms in vivo. J. Natl Cancer Inst. 1995;87:902–907. - PubMed
-
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
