

Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles
- PMID: 24336048
- PMCID: PMC3950321
- DOI: 10.1038/cdd.2013.180
Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles
Abstract
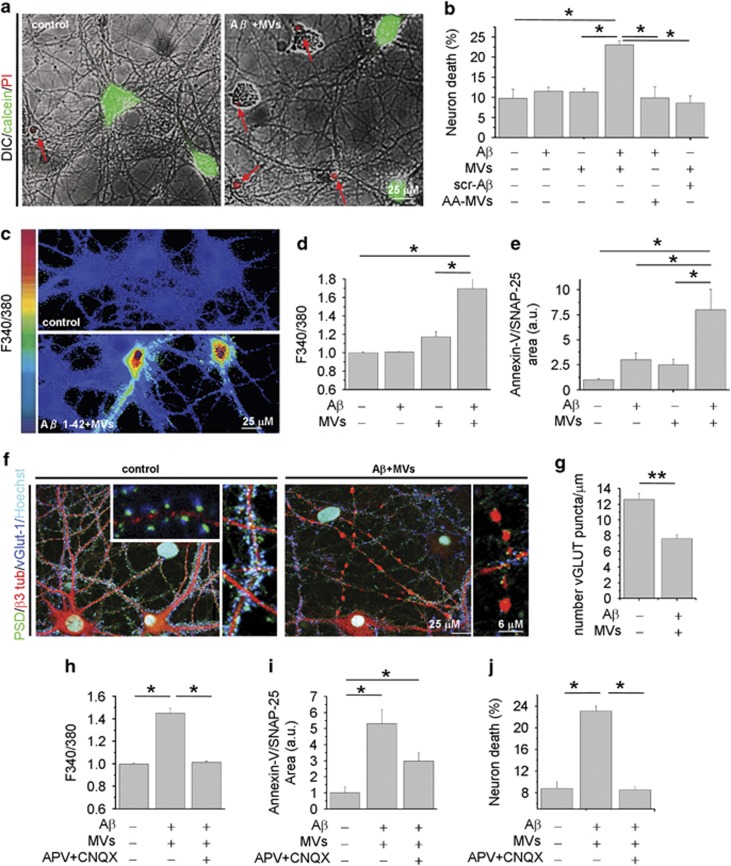
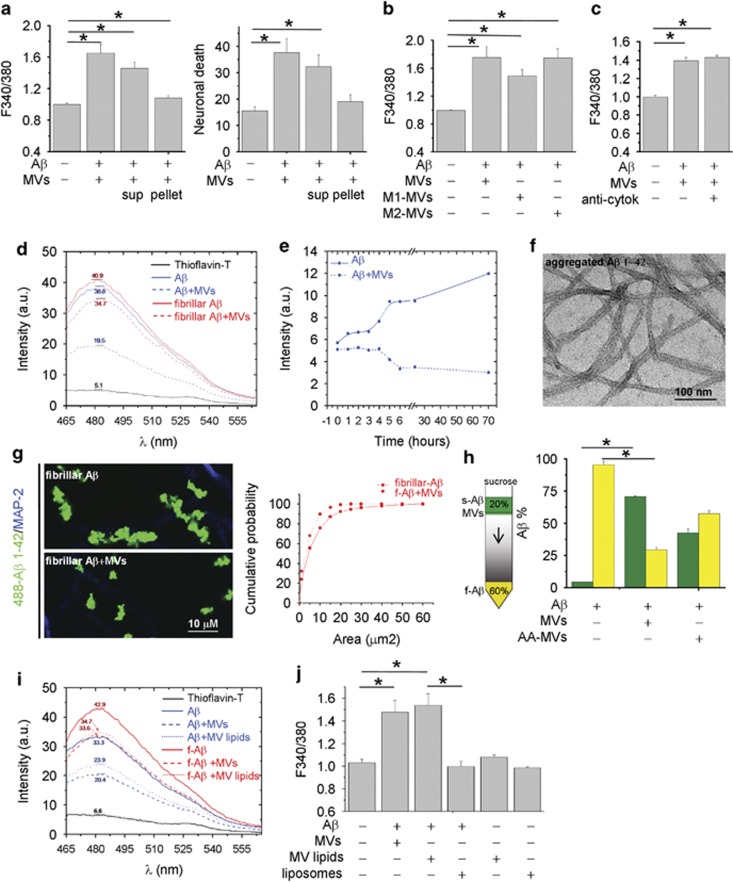
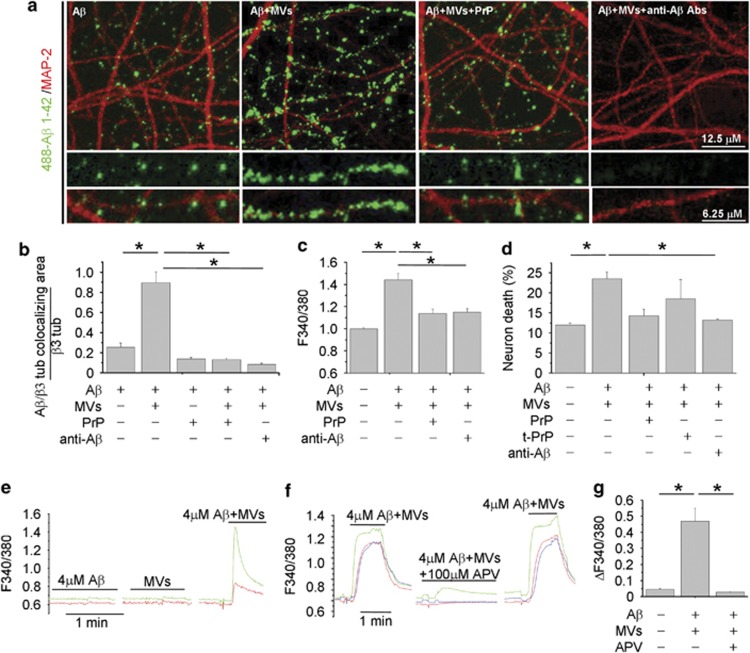
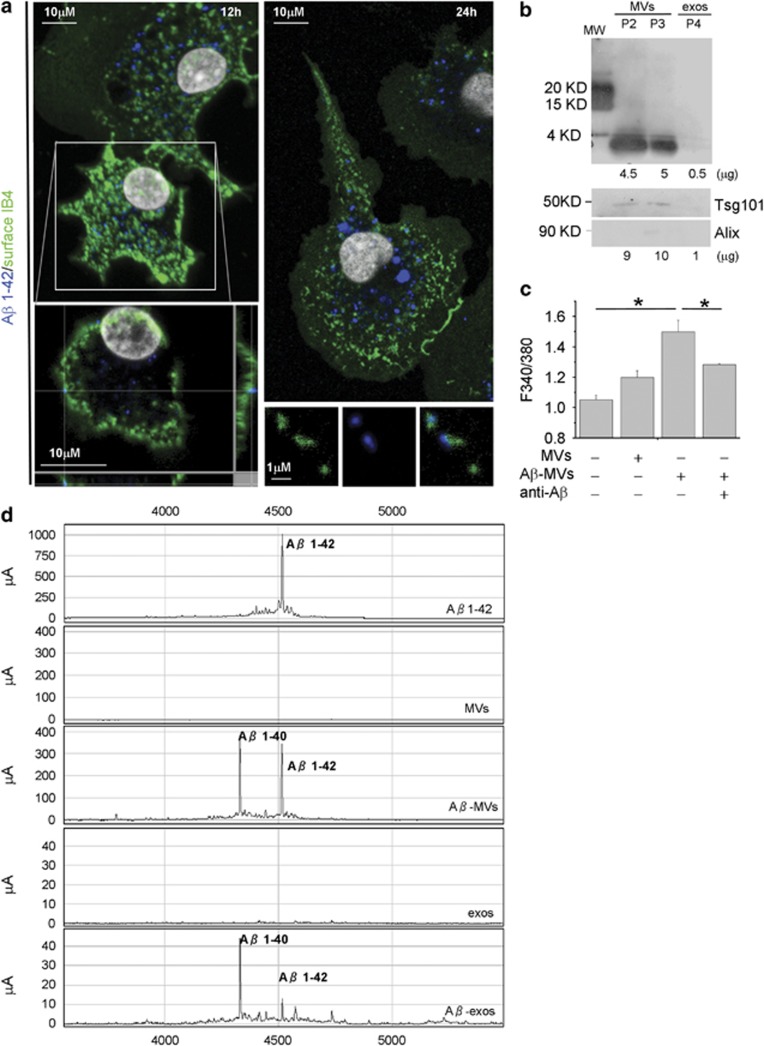
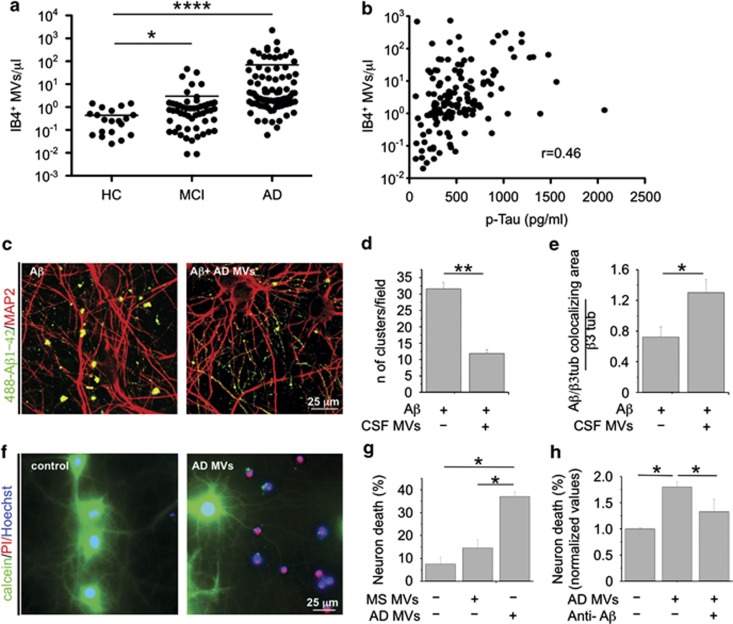
Alzheimer's disease (AD) is characterized by extracellular amyloid-β (Aβ) deposition, which activates microglia, induces neuroinflammation and drives neurodegeneration. Recent evidence indicates that soluble pre-fibrillar Aβ species, rather than insoluble fibrils, are the most toxic forms of Aβ. Preventing soluble Aβ formation represents, therefore, a major goal in AD. We investigated whether microvesicles (MVs) released extracellularly by reactive microglia may contribute to AD degeneration. We found that production of myeloid MVs, likely of microglial origin, is strikingly high in AD patients and in subjects with mild cognitive impairment and that AD MVs are toxic for cultured neurons. The mechanism responsible for MV neurotoxicity was defined in vitro using MVs produced by primary microglia. We demonstrated that neurotoxicity of MVs results from (i) the capability of MV lipids to promote formation of soluble Aβ species from extracellular insoluble aggregates and (ii) from the presence of neurotoxic Aβ forms trafficked to MVs after Aβ internalization into microglia. MV neurotoxicity was neutralized by the Aβ-interacting protein PrP and anti-Aβ antibodies, which prevented binding to neurons of neurotoxic soluble Aβ species. This study identifies microglia-derived MVs as a novel mechanism by which microglia participate in AD degeneration, and suggest new therapeutic strategies for the treatment of the disease.
Figures
Similar articles
-
Aβ42 Protofibrils Interact with and Are Trafficked through Microglial-Derived Microvesicles.ACS Chem Neurosci. 2018 Jun 20;9(6):1416-1425. doi: 10.1021/acschemneuro.8b00029. Epub 2018 Mar 22. ACS Chem Neurosci. 2018. PMID: 29543435
-
Neuroprotective effect of Chunghyuldan from amyloid beta oligomer induced neuroinflammation in vitro and in vivo.Can J Physiol Pharmacol. 2014 Jun;92(6):429-37. doi: 10.1139/cjpp-2013-0229. Epub 2014 Mar 26. Can J Physiol Pharmacol. 2014. PMID: 24820962
-
System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40.J Neurosci. 2006 Mar 22;26(12):3345-56. doi: 10.1523/JNEUROSCI.5186-05.2006. J Neurosci. 2006. PMID: 16554485 Free PMC article.
-
Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer's disease.Neurol Res. 2005 Dec;27(8):869-81. doi: 10.1179/016164105X49436. Neurol Res. 2005. PMID: 16354549 Review.
-
Amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review.Free Radic Res. 2002 Dec;36(12):1307-13. doi: 10.1080/1071576021000049890. Free Radic Res. 2002. PMID: 12607822 Review.
Cited by
-
The P2X7 Receptor: Central Hub of Brain Diseases.Front Mol Neurosci. 2020 Jul 31;13:124. doi: 10.3389/fnmol.2020.00124. eCollection 2020. Front Mol Neurosci. 2020. PMID: 32848594 Free PMC article. Review.
-
The multiple faces of extracellular vesicles released by microglia: Where are we 10 years after?Front Cell Neurosci. 2022 Sep 13;16:984690. doi: 10.3389/fncel.2022.984690. eCollection 2022. Front Cell Neurosci. 2022. PMID: 36176630 Free PMC article. Review.
-
Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death.Metab Brain Dis. 2020 Jan;35(1):11-30. doi: 10.1007/s11011-019-00516-y. Epub 2019 Dec 6. Metab Brain Dis. 2020. PMID: 31811496 Review.
-
Review on the roles of specific cell-derived exosomes in Alzheimer's disease.Front Neurosci. 2022 Jul 28;16:936760. doi: 10.3389/fnins.2022.936760. eCollection 2022. Front Neurosci. 2022. PMID: 35968378 Free PMC article. Review.
-
Role of Glia-Derived Extracellular Vesicles in Neurodegenerative Diseases.Front Aging Neurosci. 2021 Oct 20;13:765395. doi: 10.3389/fnagi.2021.765395. eCollection 2021. Front Aging Neurosci. 2021. PMID: 34744700 Free PMC article. Review.
References
-
- Herring A, Lewejohann L, Panzer AL, Donath A, Kroll O, Sachser N, et al. Preventive and therapeutic types of environmental enrichment counteract beta amyloid pathology by different molecular mechanisms. Neurobiol Dis. 2011;42:530–538. - PubMed
-
- Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Bohm G, et al. On the seeding and oligomerization of pGlu-amyloid peptides (in vitro) Biochemistry. 2006;45:12393–12399. - PubMed
-
- Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, et al. Small-molecule conversion of toxic oligomers to nontoxic beta-sheet-rich amyloid fibrils. Nat Chem Biol. 2012;8:93–101. - PubMed
-
- Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
