

Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis
- PMID: 24337049
- PMCID: PMC3978731
- DOI: 10.1161/CIRCRESAHA.114.302414
Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis
Abstract
Rationale: Oxidants generated by activated endothelial cells are known to induce apoptosis, a pathogenic feature of vascular injury and inflammation from multiple pathogeneses. The melastatin-family transient receptor potential 2 (TRPM2) channel is an oxidant-sensitive Ca2+ permeable channel implicated in mediating apoptosis; however, the mechanisms of gating of the supranormal Ca2+ influx required for initiating of apoptosis are not understood.
Objective: Here, we addressed the role of TRPM2 and its interaction with the short splice variant TRPM2 short variant (TRPM2-S) in mediating the Ca2+ entry burst required for induction of endothelial cell apoptosis.
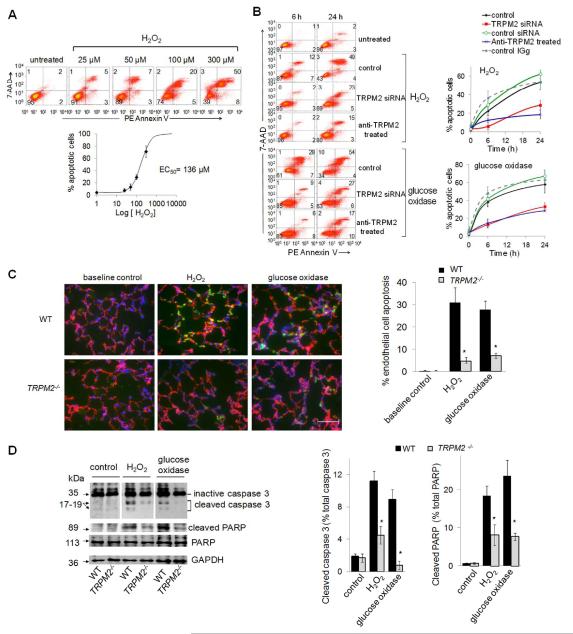
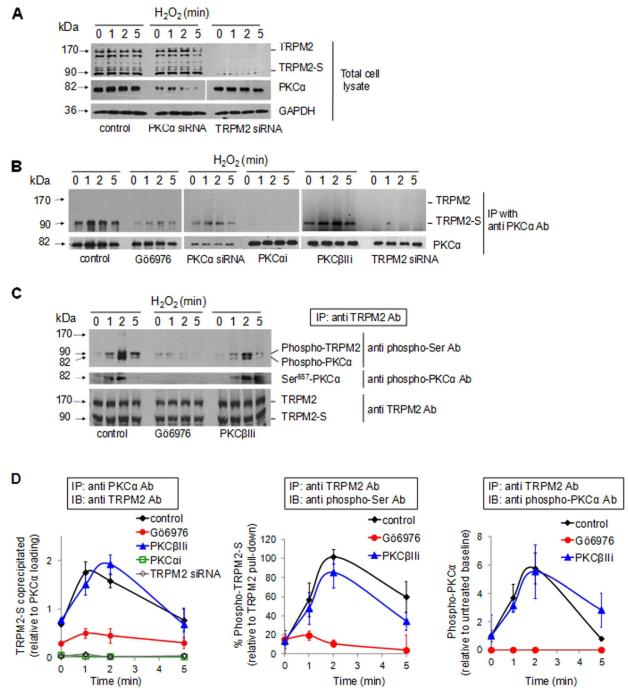
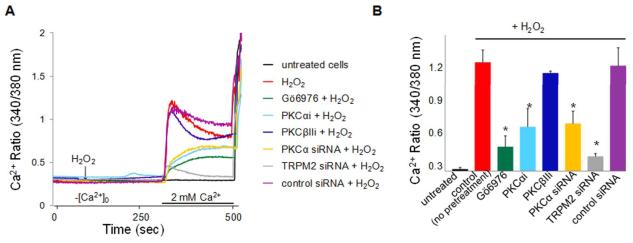
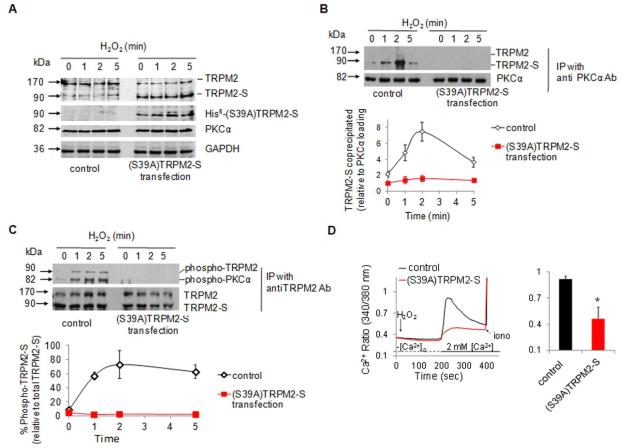
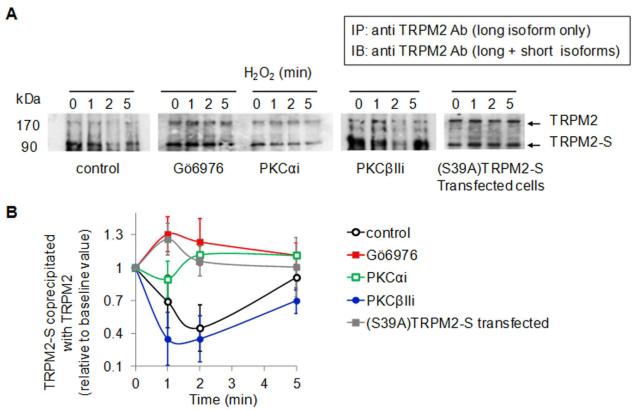
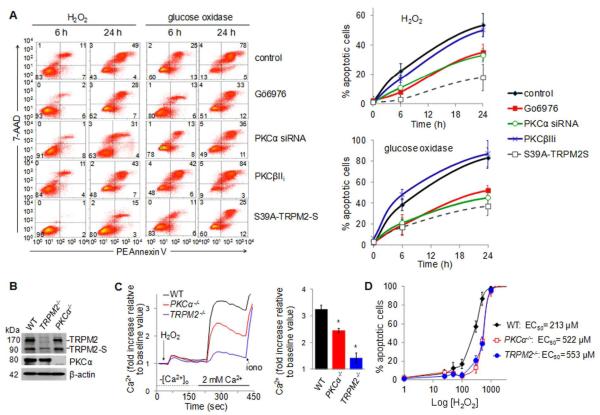
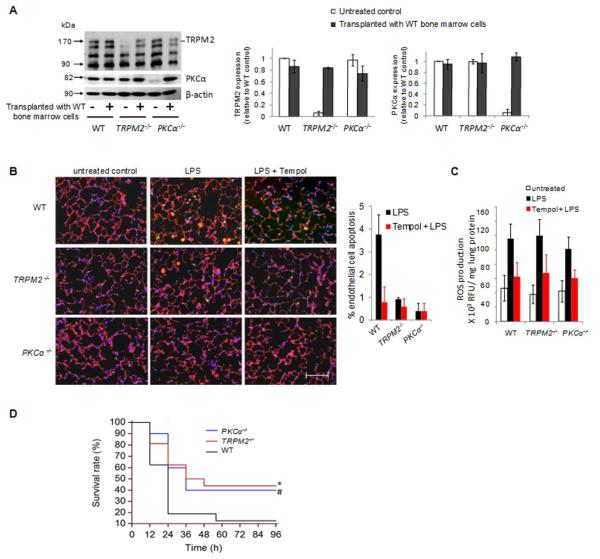
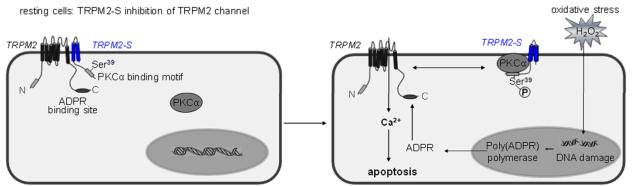
Methods and results: We observed that TRPM2-S was basally associated with TRPM2 in the endothelial plasmalemma, and this interaction functioned to suppress TRPM2-dependent Ca2+ gating constitutively. Reactive oxygen species production in endothelial cells or directly applying reactive oxygen species induced protein kinase C-α activation and phosphorylation of TRPM2 at Ser 39. This in turn stimulated a large entry of Ca2+ and activated the apoptosis pathway. A similar TRPM2-dependent endothelial apoptosis mechanism was seen in intact vessels. The protein kinase C-α-activated phosphoswitch opened the TRPM2 channel to allow large Ca2+ influx by releasing TRPM2-S inhibition of TRPM2, which in turn activated caspase-3 and cleaved the caspase substrate poly(ADP-ribose) polymerase.
Conclusions: Here, we describe a fundamental mechanism by which activation of the trp superfamily TRPM2 channel induces apoptosis of endothelial cells. The signaling mechanism involves reactive oxygen species-induced protein kinase C-α activation resulting in phosphorylation of TRPM2-S that allows enhanced TRPM2-mediated gating of Ca2+ and activation of the apoptosis program. Strategies aimed at preventing the uncoupling of TRPM2-S from TRPM2 and subsequent Ca2+ gating during oxidative stress may mitigate endothelial apoptosis and its consequences in mediating vascular injury and inflammation.
Keywords: apoptosis; capillary permeability; endothelium; inflammation.
Figures
References
-
- Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch; 2005;451:243–249. - PubMed
-
- Nazıroğlu M. TRPM2 cation channels, oxidative stress and neurological diseases: Where are we now? Neurochem Res. 2011;36:355–366. - PubMed
-
- Nagamine K, Kudoh J, Minoshima S, Kawasaki K, Asakawa S, Ito F, Shimizu N. Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics. 1998;54:124–131. - PubMed
-
- Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–C137. - PubMed
-
- Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
