

High Fat Diet Regulation of β-Cell Proliferation and β-Cell Mass
- PMID: 24339840
- PMCID: PMC3856766
- DOI: 10.2174/1874216501004010066
High Fat Diet Regulation of β-Cell Proliferation and β-Cell Mass
Abstract
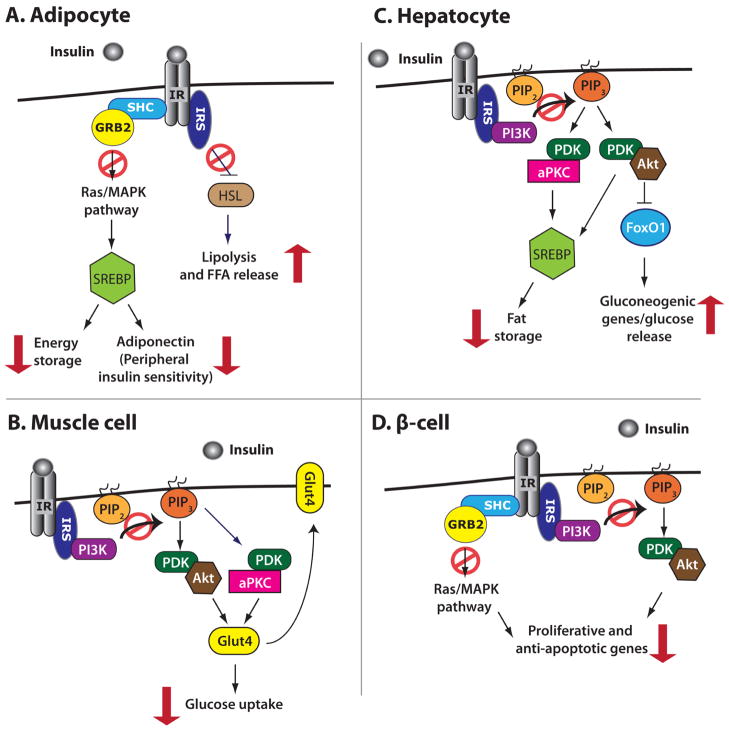
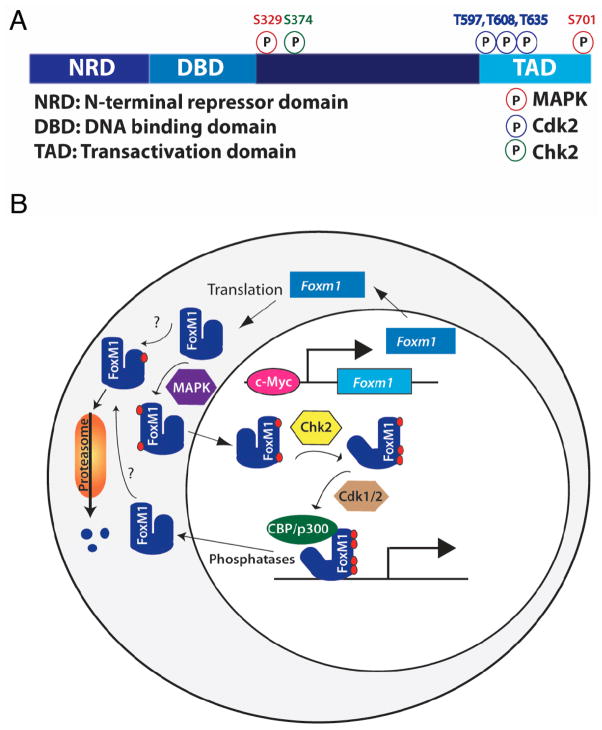
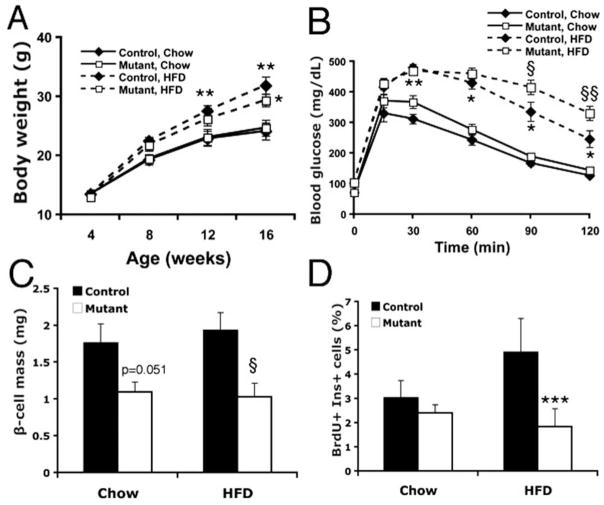
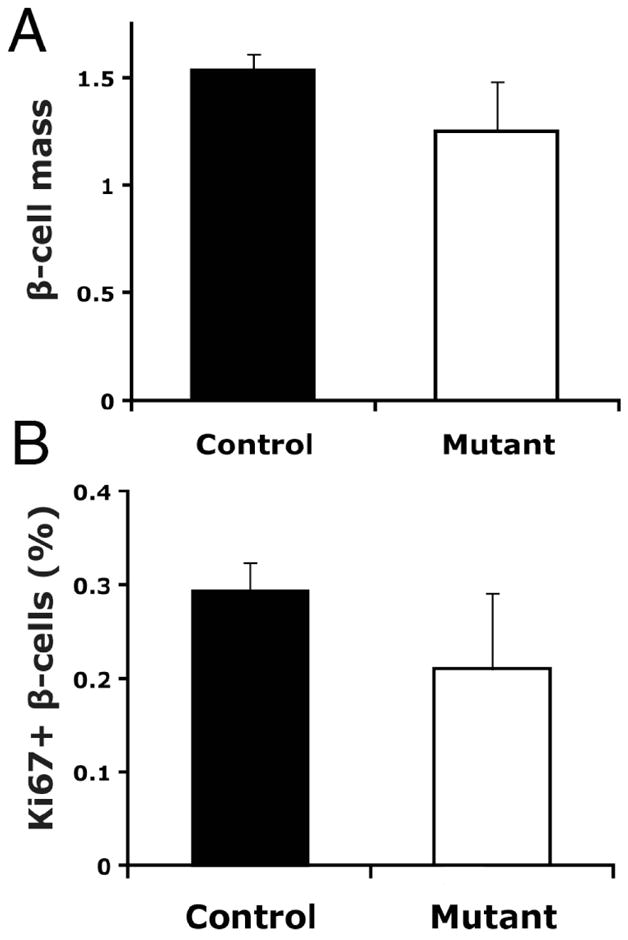
Type 2 Diabetes (T2D) is characterized by relative insulin insufficiency, caused when peripheral tissues such as liver, muscle, and adipocytes have a decreased response to insulin. One factor that elevates the risk for insulin resistance and T2D is obesity. In obese patients without T2D and initially in people who develop T2D, pancreatic β-cells are able to compensate for insulin resistance by increasing β-cell mass, effected by increased proliferation and hypertrophy, as well as increased insulin secretion per β-cell. In patients that go on to develop T2D, however, this initial period of compensation is followed by β-cell failure due to decreased proliferation and increased apoptosis. The forkhead box transcription factor FoxM1 is required for β-cell replication in mice after four weeks of age, during pregnancy, and after partial pancreatectomy. We investigated whether it is also required for β-cell proliferation due to diet-induced obesity.
Keywords: FoxM1; Obesity; diabetes; pancreas.
Figures


References
-
- Everson SA, Goldberg DE, Helmrich SP, et al. Weight gain and the risk of developing insulin resistance syndrome. Diabetes Care. 1998;21(10):1637–43. - PubMed
-
- Williamson DF. Descriptive epidemiology of body weight and weight change in U.S. adults. Ann Intern Med. 1993;1:119, 646–9. - PubMed
-
- Lewis CE, Jacobs DR, Jr, McCreath H, et al. Weight gain continues in the 1990s: 10-year trends in weight and overweight from the CARDIA study. Coronary Artery Risk Development in Young Adults. Am J Epidemiol. 2000;151(12):1172–81. - PubMed
-
- Sun XJ, Rothenberg P, Kahn CR, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352(6330):73–7. - PubMed
-
- White MF, Maron R, Kahn CR. Insulin rapidly stimulates tyrosine phosphorylation of a Mr-185,000 protein in intact cells. Nature. 1985;318(6042):183–6. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous