

Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique
- PMID: 24341750
- PMCID: PMC3890552
- DOI: 10.1186/1471-2164-14-888
Comprehensive analysis of the Corynebacterium glutamicum transcriptome using an improved RNAseq technique
Abstract
Background: The use of RNAseq to resolve the transcriptional organization of an organism was established in recent years and also showed the complexity and dynamics of bacterial transcriptomes. The aim of this study was to comprehensively investigate the transcriptome of the industrially relevant amino acid producer and model organism Corynebacterium glutamicum by RNAseq in order to improve its genome annotation and to describe important features for transcription and translation.
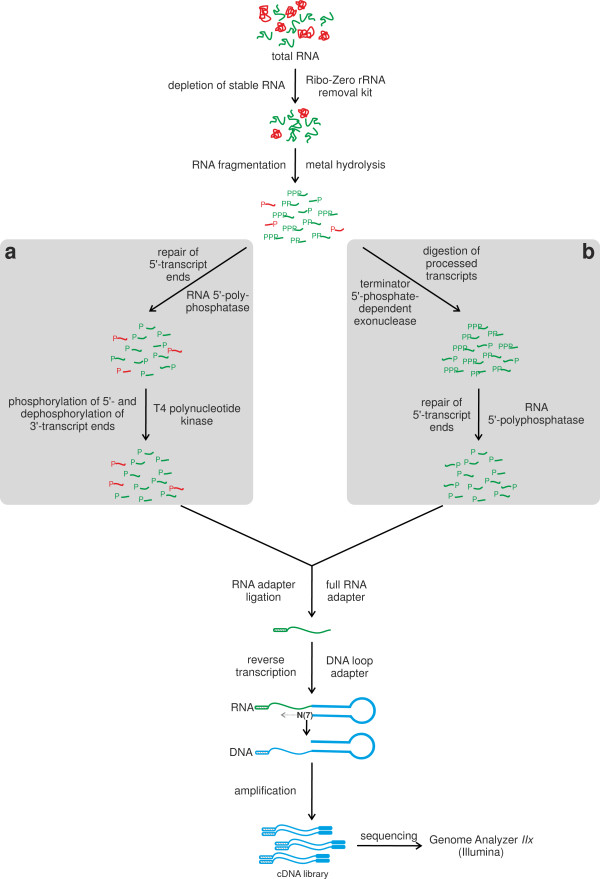
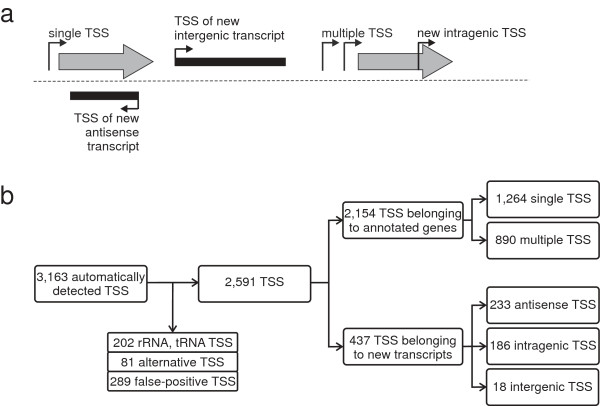
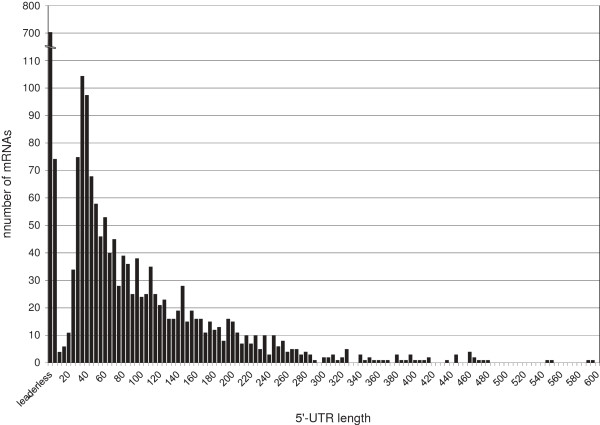
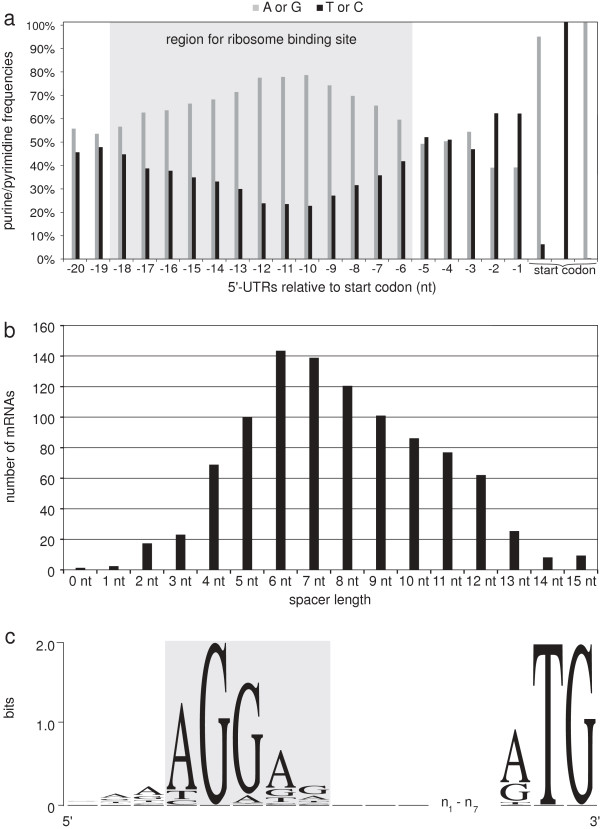
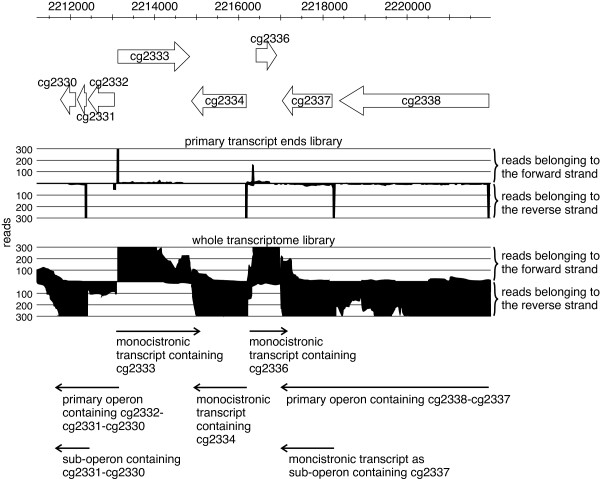
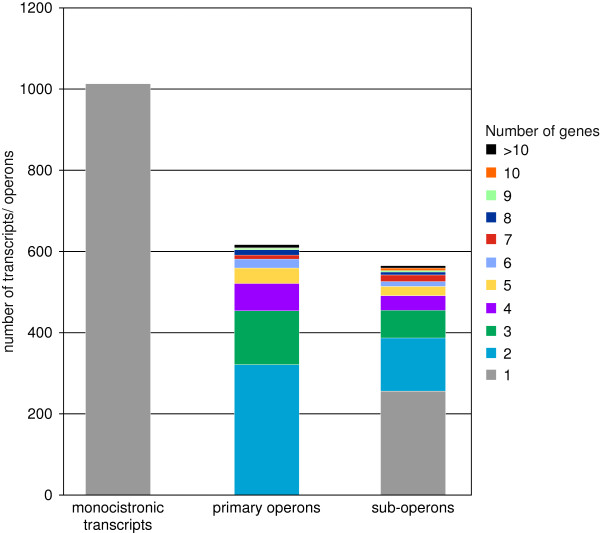
Results: RNAseq data sets were obtained by two methods, one that focuses on 5'-ends of primary transcripts and another that provides the overall transcriptome with an improved resolution of 3'-ends of transcripts. Subsequent data analysis led to the identification of more than 2,000 transcription start sites (TSSs), the definition of 5'-UTRs (untranslated regions) for annotated protein-coding genes, operon structures and many novel transcripts located between or in antisense orientation to protein-coding regions. Interestingly, a high number of mRNAs (33%) is transcribed as leaderless transcripts. From the data, consensus promoter and ribosome binding site (RBS) motifs were identified and it was shown that the majority of genes in C. glutamicum are transcribed monocistronically, but operons containing up to 16 genes are also present.
Conclusions: The comprehensive transcriptome map of C. glutamicum established in this study represents a major step forward towards a complete definition of genetic elements (e.g. promoter regions, gene starts and stops, 5'-UTRs, RBSs, transcript starts and ends) and provides the ideal basis for further analyses on transcriptional regulatory networks in this organism. The methods developed are easily applicable for other bacteria and have the potential to be used also for quantification of transcriptomes, replacing microarrays in the near future.
Figures
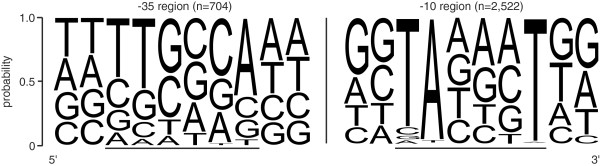

Similar articles
-
Transcriptome analysis of thermophilic methylotrophic Bacillus methanolicus MGA3 using RNA-sequencing provides detailed insights into its previously uncharted transcriptional landscape.BMC Genomics. 2015 Feb 14;16(1):73. doi: 10.1186/s12864-015-1239-4. BMC Genomics. 2015. PMID: 25758049 Free PMC article.
-
RNAseq analysis of α-proteobacterium Gluconobacter oxydans 621H.BMC Genomics. 2018 Jan 6;19(1):24. doi: 10.1186/s12864-017-4415-x. BMC Genomics. 2018. PMID: 29304737 Free PMC article.
-
Genome-wide determination of transcription start sites reveals new insights into promoter structures in the actinomycete Corynebacterium glutamicum.J Biotechnol. 2017 Sep 10;257:99-109. doi: 10.1016/j.jbiotec.2017.04.008. Epub 2017 Apr 13. J Biotechnol. 2017. PMID: 28412515
-
Corynebacterium glutamicum promoters: a practical approach.Microb Biotechnol. 2013 Mar;6(2):103-17. doi: 10.1111/1751-7915.12019. Epub 2013 Jan 10. Microb Biotechnol. 2013. PMID: 23305350 Free PMC article. Review.
-
Deep sequencing approaches for the analysis of prokaryotic transcriptional boundaries and dynamics.Methods. 2017 May 1;120:76-84. doi: 10.1016/j.ymeth.2017.04.016. Epub 2017 Apr 21. Methods. 2017. PMID: 28434904 Review.
Cited by
-
A universal approach to gene expression engineering.Synth Biol (Oxf). 2022 Aug 22;7(1):ysac017. doi: 10.1093/synbio/ysac017. eCollection 2022. Synth Biol (Oxf). 2022. PMID: 36212995 Free PMC article.
-
ReadXplorer 2-detailed read mapping analysis and visualization from one single source.Bioinformatics. 2016 Dec 15;32(24):3702-3708. doi: 10.1093/bioinformatics/btw541. Epub 2016 Aug 18. Bioinformatics. 2016. PMID: 27540267 Free PMC article.
-
Biosensor-based isolation of amino acid-producing Vibrio natriegens strains.Metab Eng Commun. 2021 Nov 11;13:e00187. doi: 10.1016/j.mec.2021.e00187. eCollection 2021 Dec. Metab Eng Commun. 2021. PMID: 34824977 Free PMC article.
-
Overlap of Promoter Recognition Specificity of Stress Response Sigma Factors SigD and SigH in Corynebacterium glutamicum ATCC 13032.Front Microbiol. 2019 Jan 9;9:3287. doi: 10.3389/fmicb.2018.03287. eCollection 2018. Front Microbiol. 2019. PMID: 30687273 Free PMC article.
-
OsnR is an autoregulatory negative transcription factor controlling redox-dependent stress responses in Corynebacterium glutamicum.Microb Cell Fact. 2021 Oct 18;20(1):203. doi: 10.1186/s12934-021-01693-1. Microb Cell Fact. 2021. PMID: 34663317 Free PMC article.
References
-
- Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Krämer R, Linke B, McHardy AC, Meyer F, Möckel B, Pfefferle W, Pühler A, Rey DA, Rückert C, Rupp O, Sahm H, Wendisch VF, Wiegräbe I, Tauch A. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003;14:5–25. doi: 10.1016/S0168-1656(03)00154-8. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
