

Autophagy regulation by nutrient signaling
- PMID: 24343578
- PMCID: PMC3879708
- DOI: 10.1038/cr.2013.166
Autophagy regulation by nutrient signaling
Abstract
The ability of cells to respond to changes in nutrient availability is essential for the maintenance of metabolic homeostasis and viability. One of the key cellular responses to nutrient withdrawal is the upregulation of autophagy. Recently, there has been a rapid expansion in our knowledge of the molecular mechanisms involved in the regulation of mammalian autophagy induction in response to depletion of key nutrients. Intracellular amino acids, ATP, and oxygen levels are intimately tied to the cellular balance of anabolic and catabolic processes. Signaling from key nutrient-sensitive kinases mTORC1 and AMP-activated protein kinase (AMPK) is essential for the nutrient sensing of the autophagy pathway. Recent advances have shown that the nutrient status of the cell is largely passed on to the autophagic machinery through the coordinated regulation of the ULK and VPS34 kinase complexes. Identification of extensive crosstalk and feedback loops converging on the regulation of ULK and VPS34 can be attributed to the importance of these kinases in autophagy induction and maintaining cellular homeostasis.
Figures
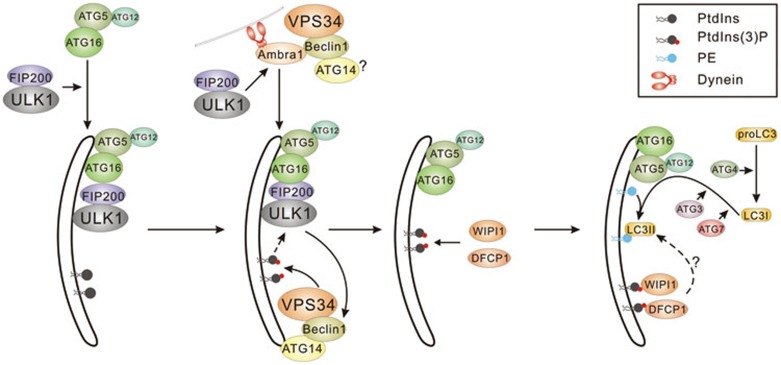
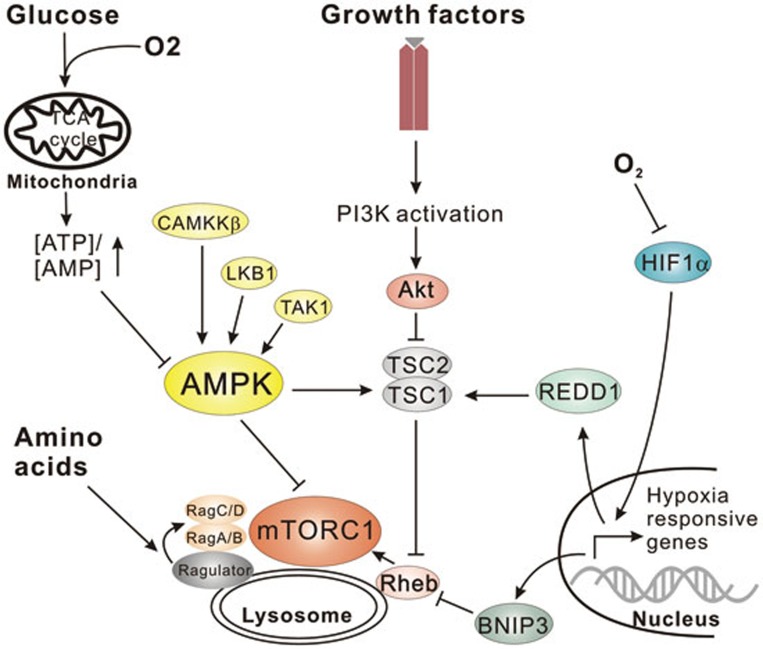
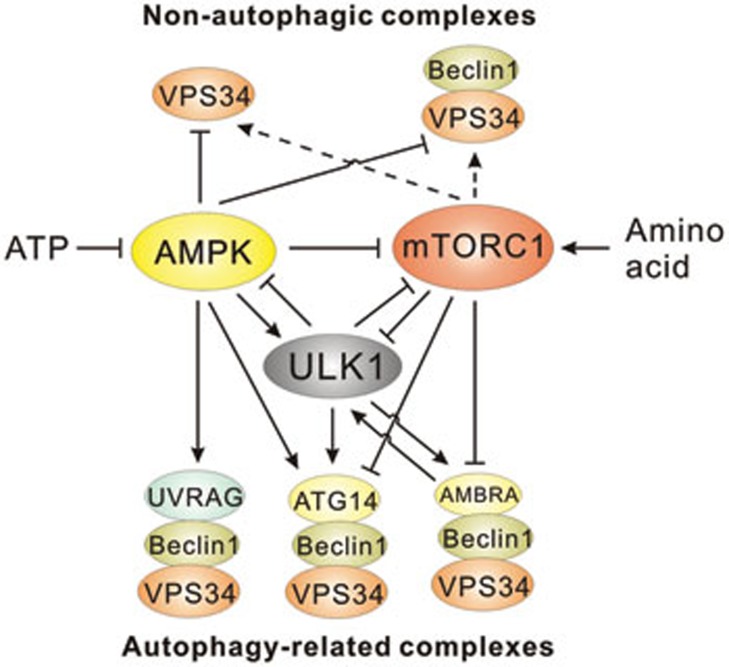
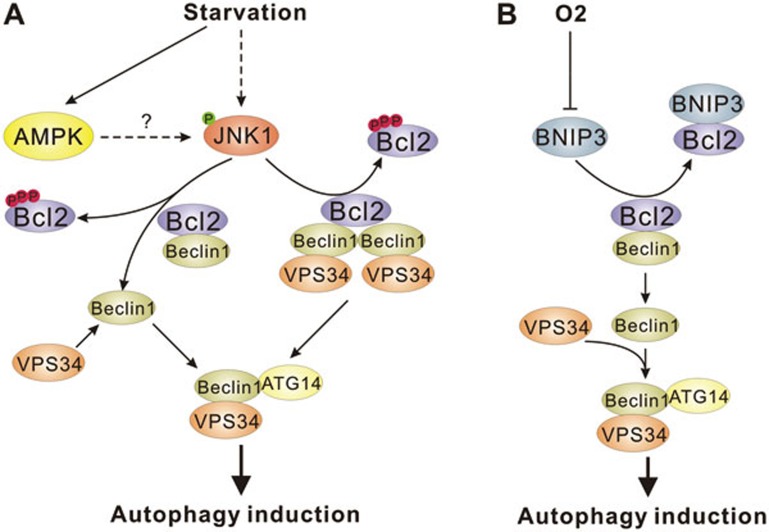
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
