

Interpretation of genetic variants
- PMID: 24343785
- PMCID: PMC4364415
- DOI: 10.1136/thoraxjnl-2013-204903
Interpretation of genetic variants
Abstract
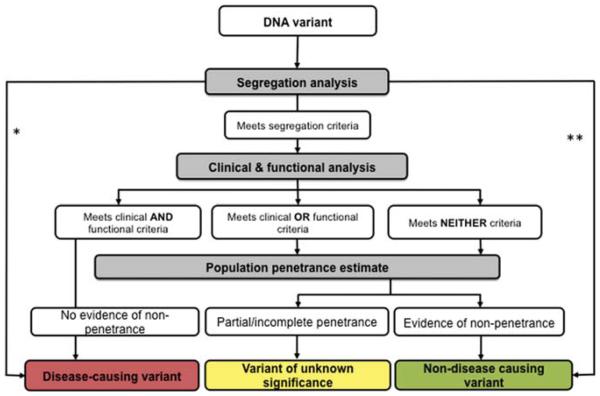
Sequencing of the human genome and introduction of clinical next-generation sequencing enable discovery of all DNA variants carried by an individual. Variants may be solely responsible for disease, may contribute to disease, or may have no influence on the development of disease. Interpreting the effect of these variants upon disease is a major challenge for medicine. Although the process is still evolving, certain methods are useful in discriminating the effect of variants upon phenotype. These methods have been employed to the greatest extent in Mendelian disorders where deleterious changes in one gene can cause disease. Here, we briefly review the relative merits of these methods, with emphasis on using a comprehensive approach modelled after the analysis of variants that causes cystic fibrosis.
Keywords: Genetic Variation; Genotype; Mutation Analysis; Phenotype; Population Genetics.
Figures
References
-
- Lane KB, Machado RD, Pauciulo MW, et al. International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. - PubMed
-
- Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical