

The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin
- PMID: 24344137
- PMCID: PMC3931073
- DOI: 10.1074/jbc.M113.511204
The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac β-myosin
Abstract
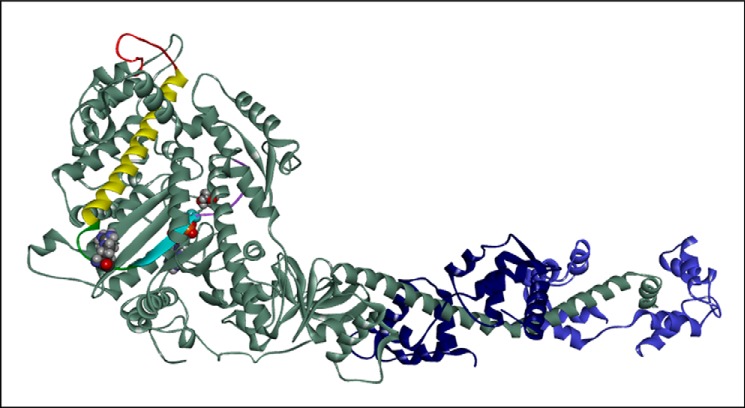
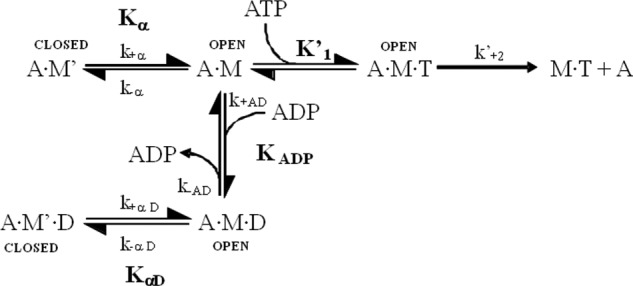
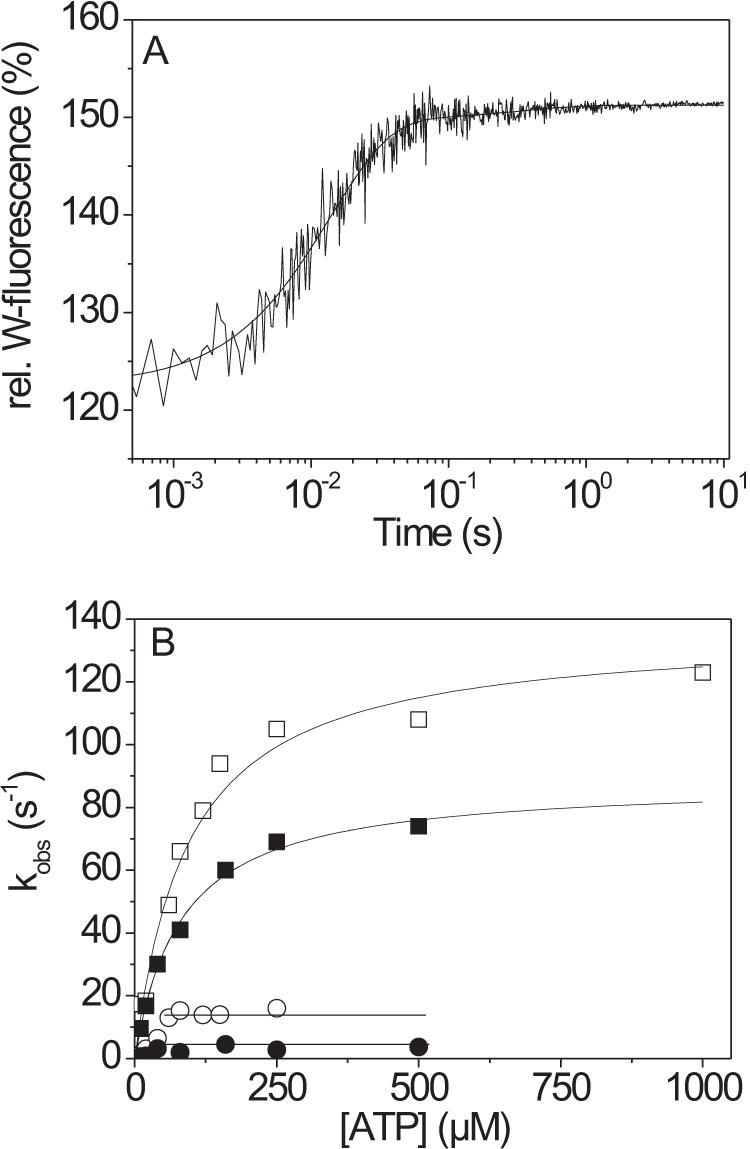
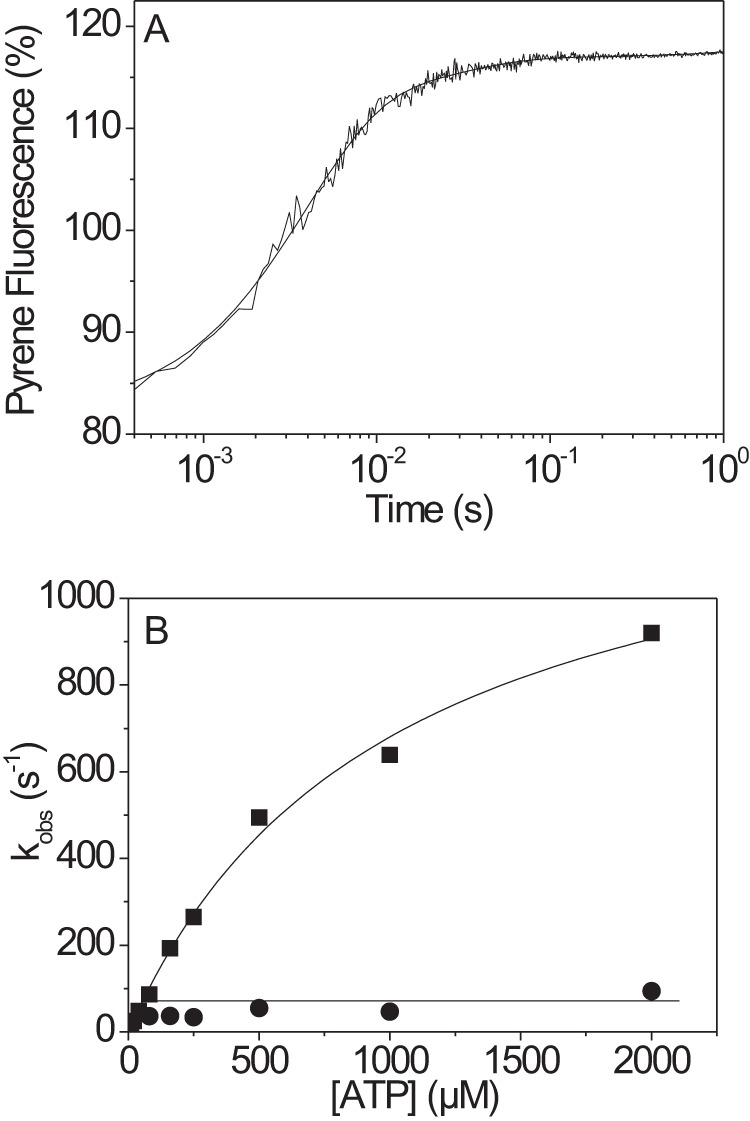
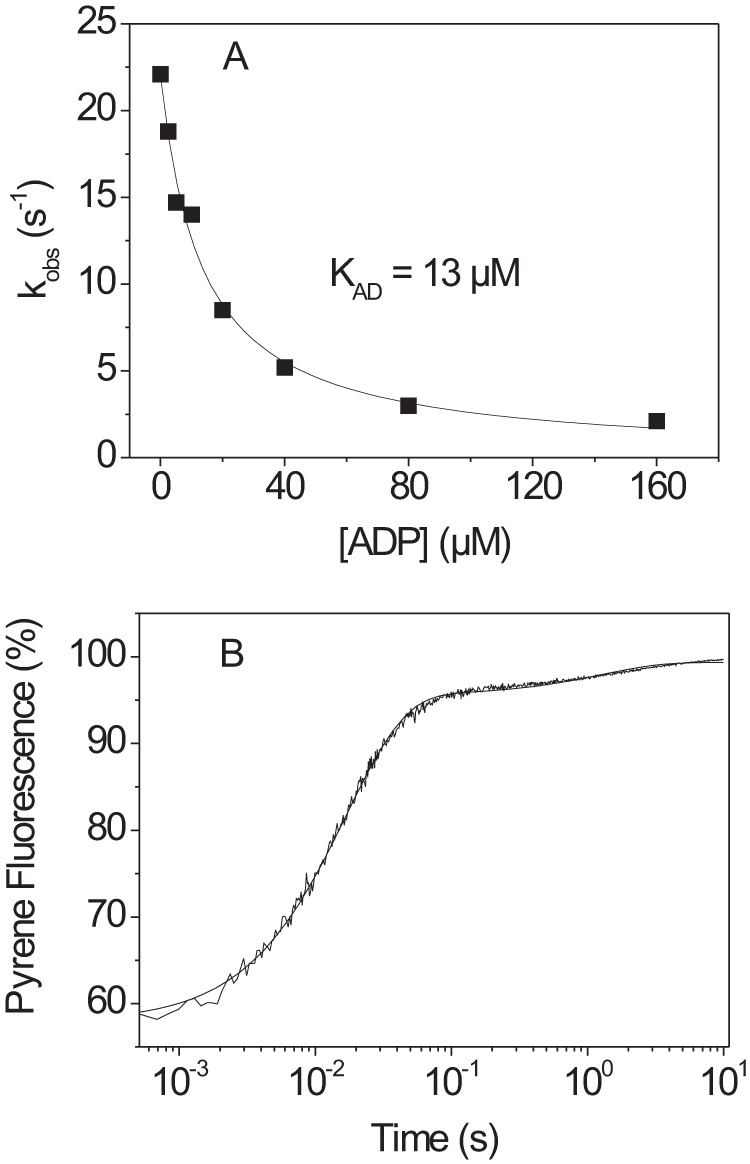
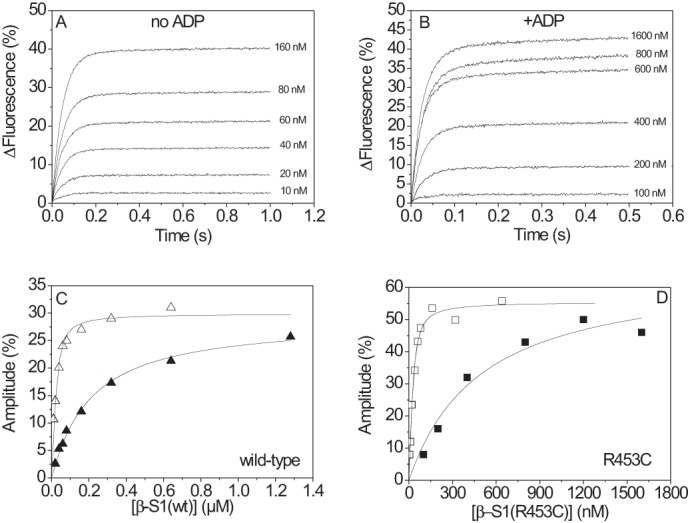
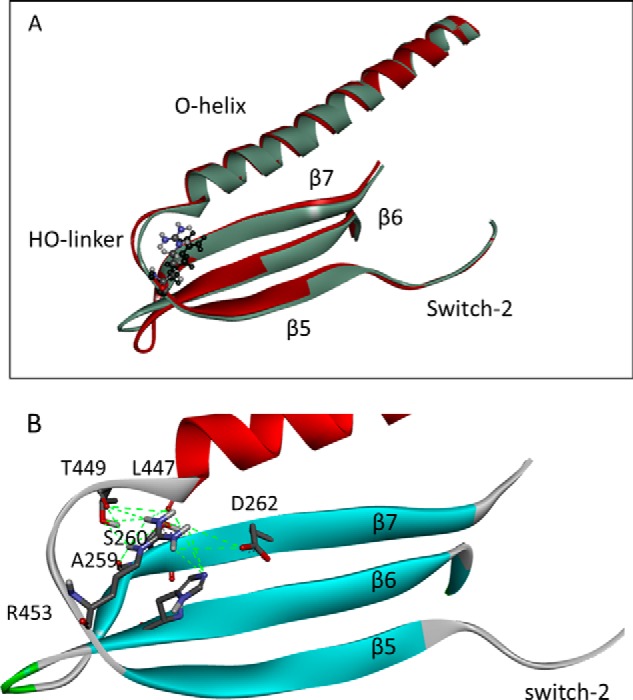
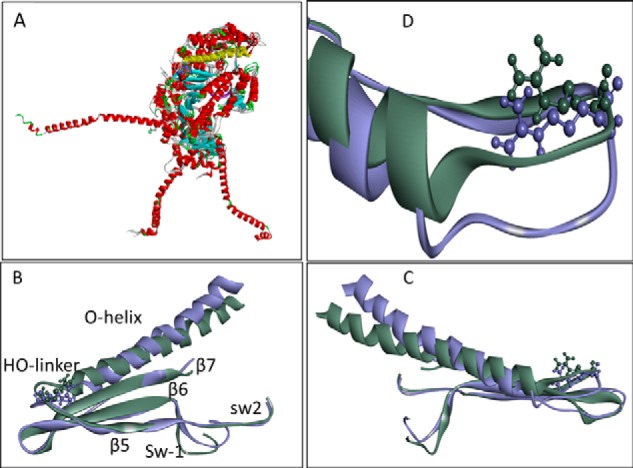
The human hypertrophic cardiomyopathy mutation R453C results in one of the more severe forms of the myopathy. Arg-453 is found in a conserved surface loop of the upper 50-kDa domain of the myosin motor domain and lies between the nucleotide binding pocket and the actin binding site. It connects to the cardiomyopathy loop via a long α-helix, helix O, and to Switch-2 via the fifth strand of the central β-sheet. The mutation is, therefore, in a position to perturb a wide range of myosin molecular activities. We report here the first detailed biochemical kinetic analysis of the motor domain of the human β-cardiac myosin carrying the R453C mutation. A recent report of the same mutation (Sommese, R. F., Sung, J., Nag, S., Sutton, S., Deacon, J. C., Choe, E., Leinwand, L. A., Ruppel, K., and Spudich, J. A. (2013) Proc. Natl. Acad. Sci. U.S.A. 110, 12607-12612) found reduced ATPase and in vitro motility but increased force production using an optical trap. Surprisingly, our results show that the mutation alters few biochemical kinetic parameters significantly. The exceptions are the rate constants for ATP binding to the motor domain (reduced by 35%) and the ATP hydrolysis step/recovery stroke (slowed 3-fold), which could be the rate-limiting step for the ATPase cycle. Effects of the mutation on the recovery stroke are consistent with a perturbation of Switch-2 closure, which is required for the recovery stroke and the subsequent ATP hydrolysis.
Keywords: Actin; Cardiac Muscle; Cardiomyopathy; Fluorescence; Homology Models; Kinetics; Myosin; Protein Structure-Function; Sequence Alignment.
Figures

References
-
- Liew C. C., Dzau V. J. (2004) Molecular genetics and genomics of heart failure. Nat. Rev. Genet. 5, 811–825 - PubMed
-
- Ramaraj R. (2008) Hypertrophic cardiomyopathy. Etiology, diagnosis, and treatment. Cardiol. Rev. 16, 172–180 - PubMed
-
- Maron B. J. (2002) Hypertrophic cardiomyopathy. Circulation 106, 2419–2421 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
