

Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma
- PMID: 24346431
- PMCID: PMC3944405
- DOI: 10.1158/0008-5472.CAN-13-2800
Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma
Abstract
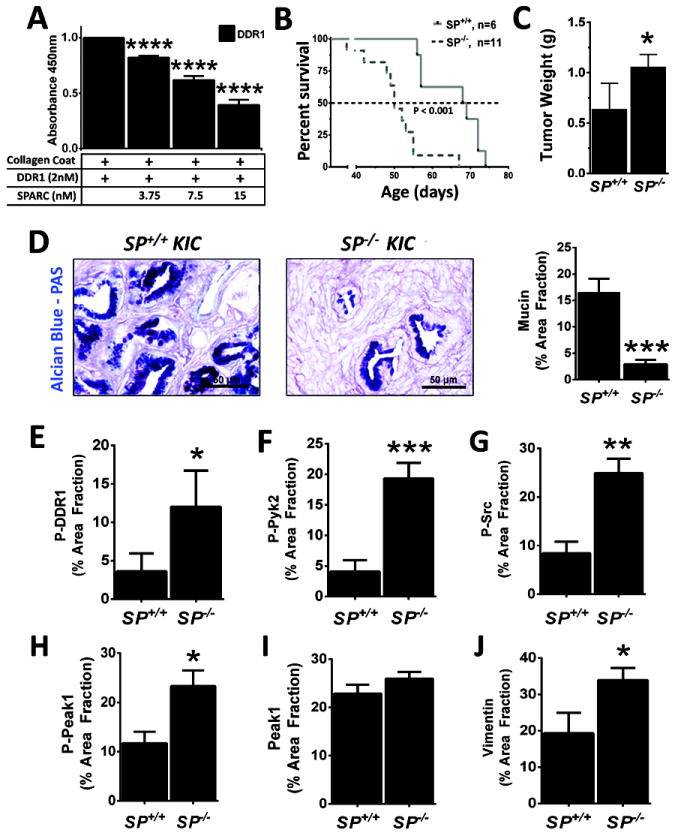
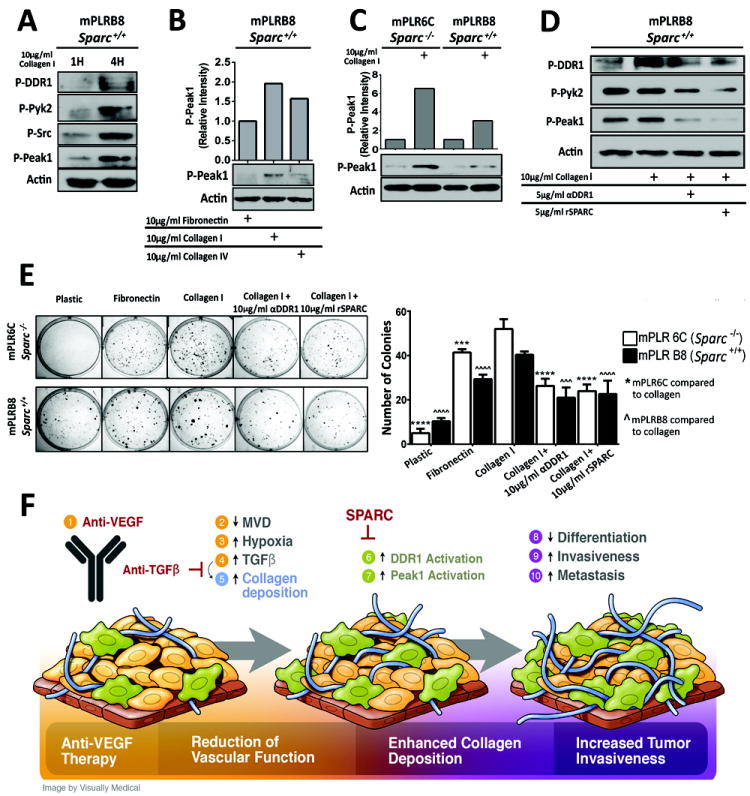
There is growing evidence that antiangiogenic therapy stimulates cancer cell invasion and metastasis. However, the underlying molecular mechanisms responsible for these changes have not been fully defined. Here, we report that anti-VEGF therapy promotes local invasion and metastasis by inducing collagen signaling in cancer cells. We show that chronic VEGF inhibition in a genetically engineered mouse model of pancreatic ductal adenocarcinoma (PDA) induces hypoxia, a less differentiated mesenchymal-like tumor cell phenotype, TGF-β expression, and collagen deposition and signaling. In addition, we show that collagen signaling is critical for protumorigenic activity of TGF-β in vitro. To further model the impact of collagen signaling in tumors, we evaluated PDA in mice lacking Sparc, a protein that reduces collagen binding to cell surface receptors. Importantly, we show that loss of Sparc increases collagen signaling and tumor progression. Together, these findings suggest that collagen actively promotes PDA spread and that enhanced disease progression associated with anti-VEGF therapy can arise from elevated extracellular matrix-mediated signaling.
©2013 AACR.
Conflict of interest statement
Figures
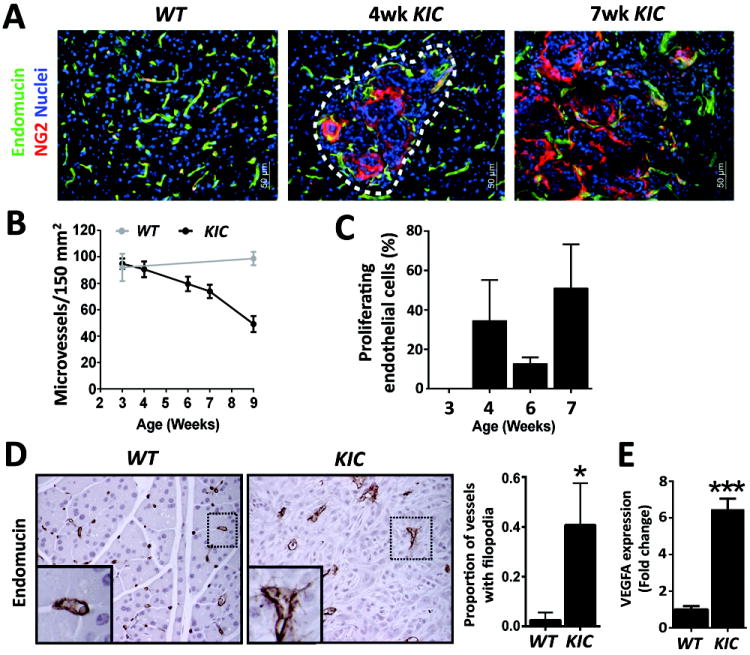
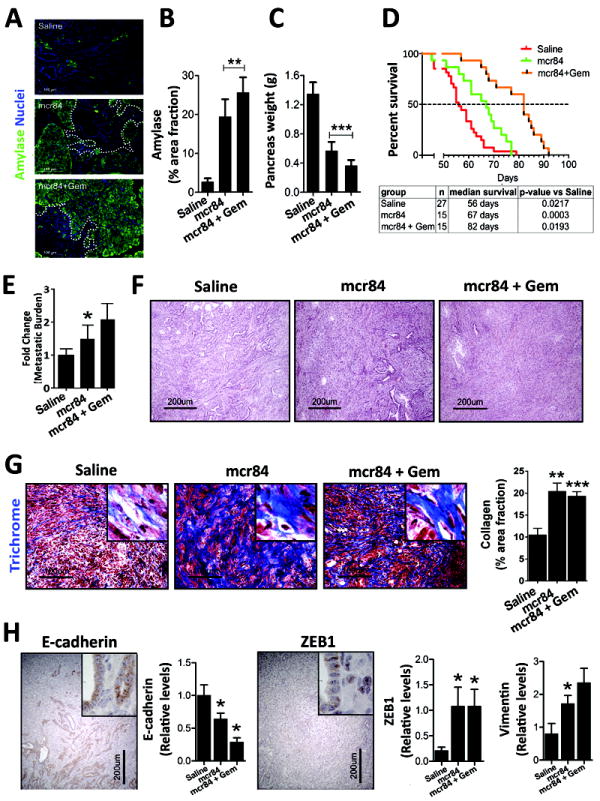
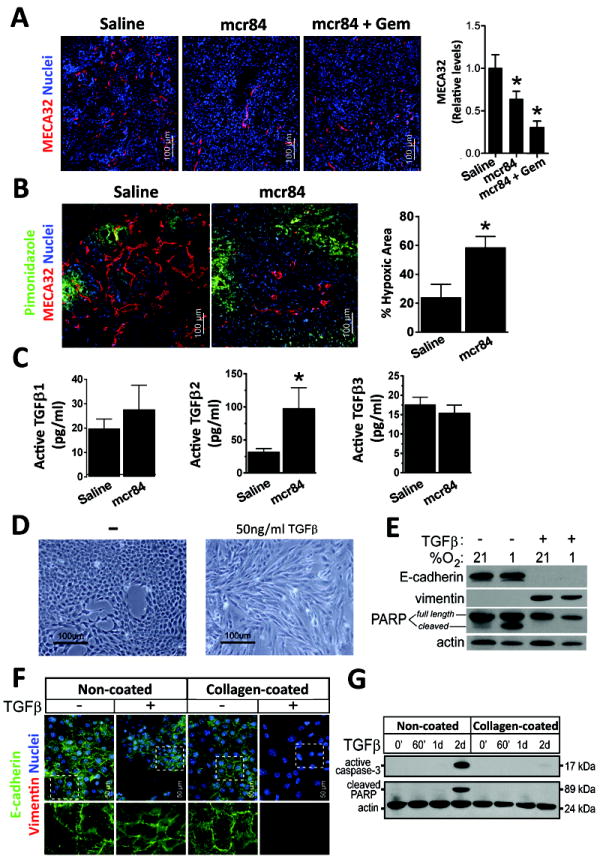
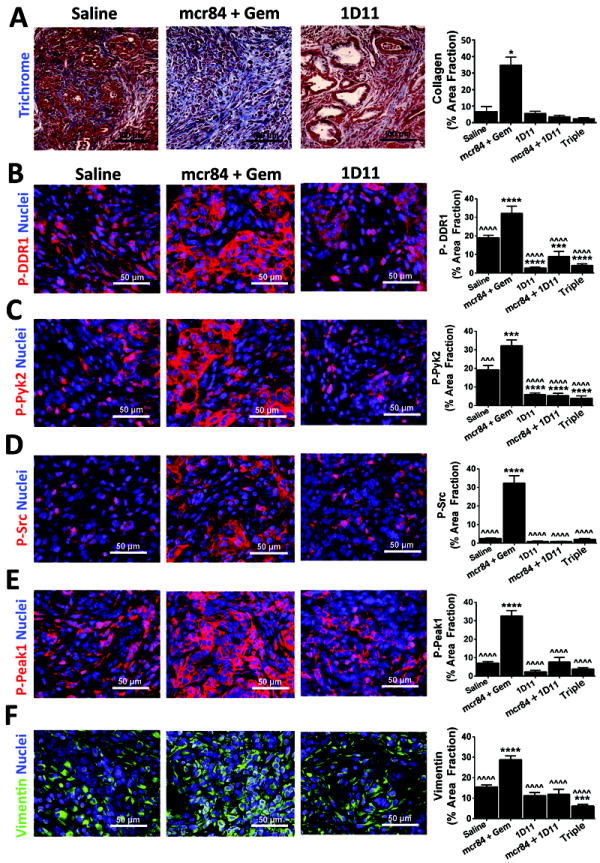
References
-
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. - PubMed
-
- Roskoski R., Jr Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62:179–213. - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. - PubMed
-
- Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320:130–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
