

Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral-like type I interferon gene response
- PMID: 24347514
- PMCID: PMC4235296
- DOI: 10.1002/ijc.28670
Comprehensive metagenomic analysis of glioblastoma reveals absence of known virus despite antiviral-like type I interferon gene response
Abstract
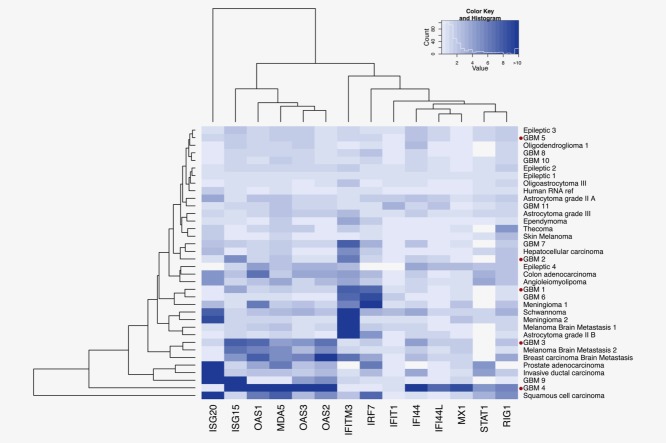
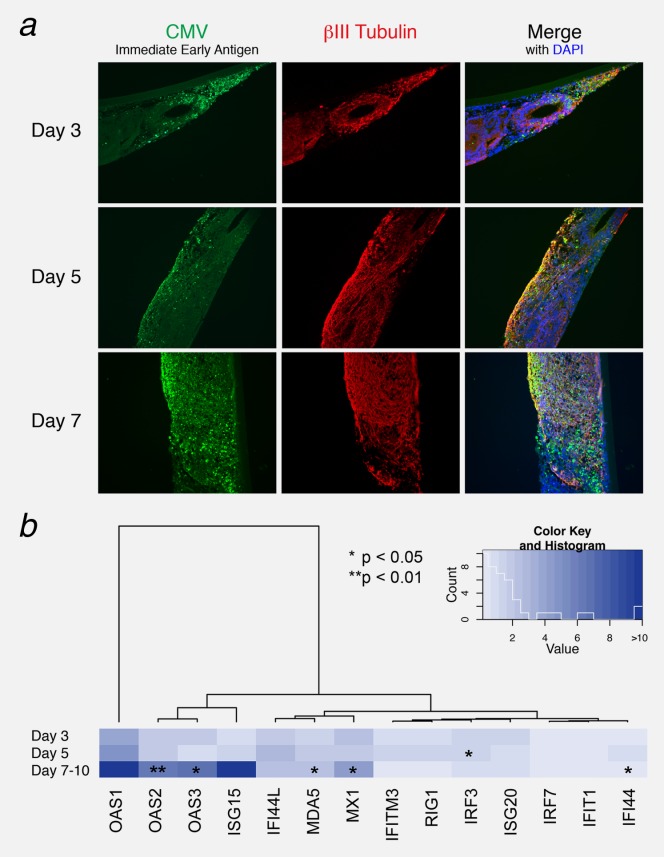
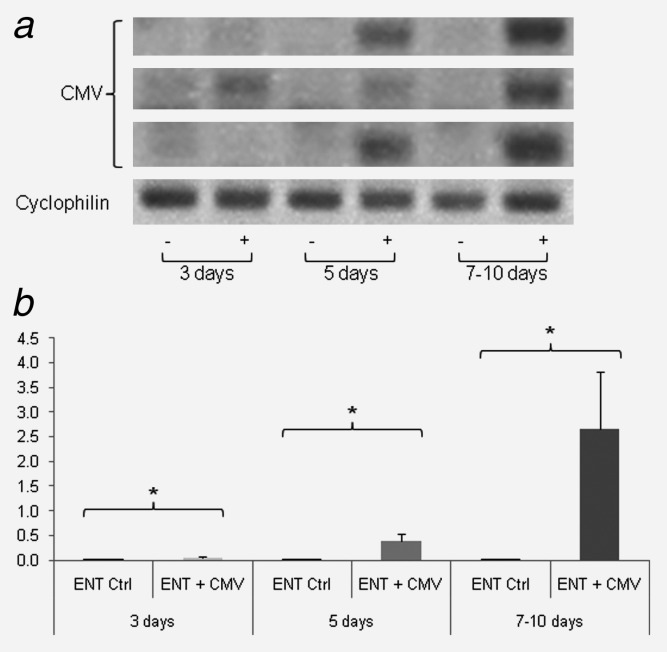
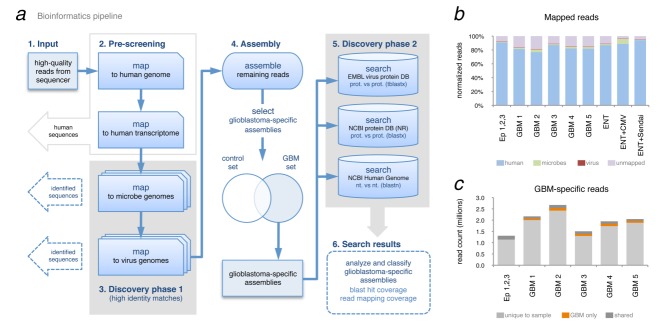
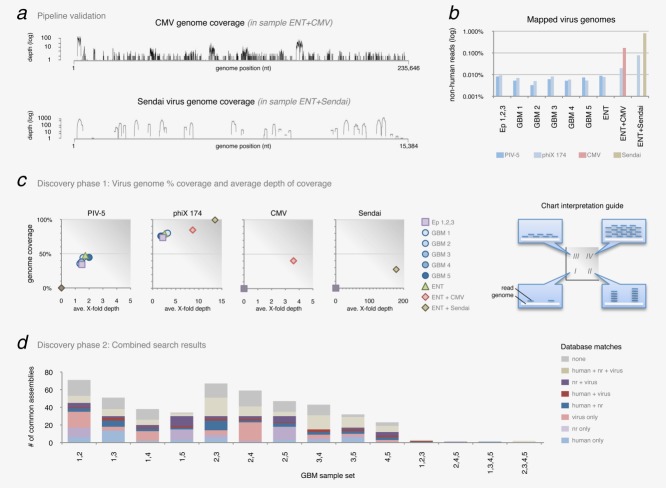
Glioblastoma is a deadly malignant brain tumor and one of the most incurable forms of cancer in need of new therapeutic targets. As some cancers are known to be caused by a virus, the discovery of viruses could open the possibility to treat, and perhaps prevent, such a disease. Although an association with viruses such as cytomegalovirus or Simian virus 40 has been strongly suggested, involvement of these and other viruses in the initiation and/or propagation of glioblastoma remains vague, controversial and warrants elucidation. To exhaustively address the association of virus and glioblastoma, we developed and validated a robust metagenomic approach to analyze patient biopsies via high-throughput sequencing, a sensitive tool for virus screening. In addition to traditional clinical diagnostics, glioblastoma biopsies were deep-sequenced and analyzed with a multistage computational pipeline to identify known or potentially discover unknown viruses. In contrast to the studies reporting the presence of viral signatures in glioblastoma, no common or recurring active viruses were detected, despite finding an antiviral-like type I interferon response in some specimens. Our findings highlight a discrete and non-specific viral signature and uncharacterized short RNA sequences in glioblastoma. This study provides new insights into glioblastoma pathogenesis and defines a general methodology that can be used for high-resolution virus screening and discovery in human cancers.
Keywords: antiviral type I interferon response; glioblastoma multiforme; high-throughput sequencing; metagenomic analysis; virus discovery.
© 2013 The Authors. Published by Wiley Periodicals, Inc. on behalf of UICC.
Figures
References
-
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. - PubMed
-
- Schwartzbaum JA, Fisher JL, Aldape KD, et al. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503; quiz 1 p following 16. - PubMed
-
- Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109:93–108. - PubMed
-
- Kouhata T, Fukuyama K, Hagihara N, et al. Detection of simian virus 40 DNA sequence in human primary glioblastomas multiforme. J Neurosurg. 2001;95:96–101. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
