

Early Detection of Huntington Disease
- PMID: 24348095
- PMCID: PMC3860286
- DOI: 10.2217/fnl.09.78
Early Detection of Huntington Disease
Abstract
Huntington disease (HD) is a devastating illness, although its autosomal dominant genetic transmission allows a unique opportunity to study apparently healthy individuals before manifest disease. Attempts to study early disease are not unique in neurology (e.g., Mild Cognitive Impairment, Vascular Cognitive Impairment), but studying otherwise-healthy appearing individuals who will go on with nearly 99% certainty to manifest the symptoms of brain disease does provide distinct but valuable information about the true natural history of the disease. The field has witnessed an explosion of research examining possible early indicators of HD during what is now referred to as the "prodrome" of HD. A NIH study in its ninth year (PREDICT-HD) has offered a glimpse into the transition from an apparently healthy state to an obviously diseased state, and can serve as a model for many other genetic diseases, both neurological and non-neurological.
Keywords: Huntington disease; biomarkers; clinical endpoints; clinical trials; detection; diagnosis; prevention.
Figures
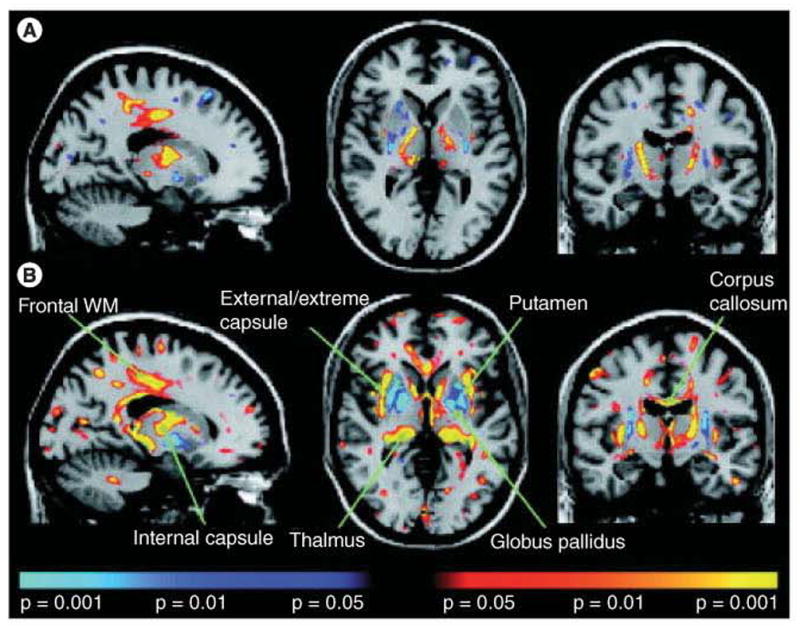
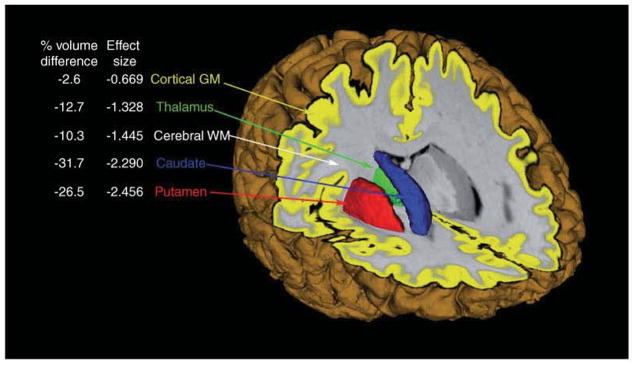
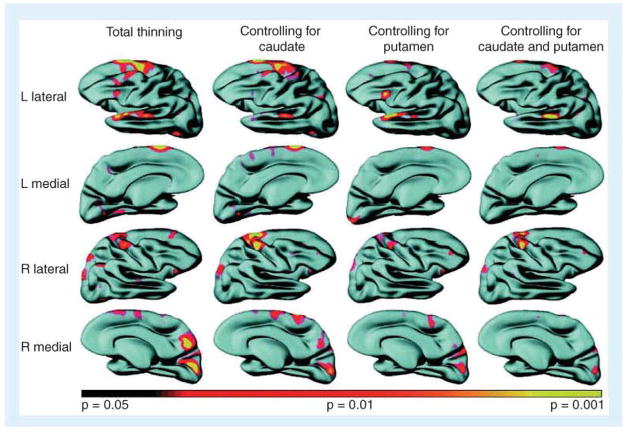
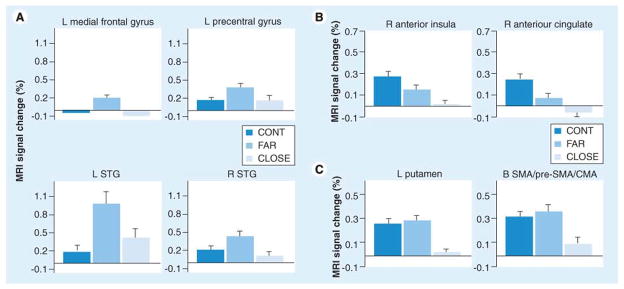
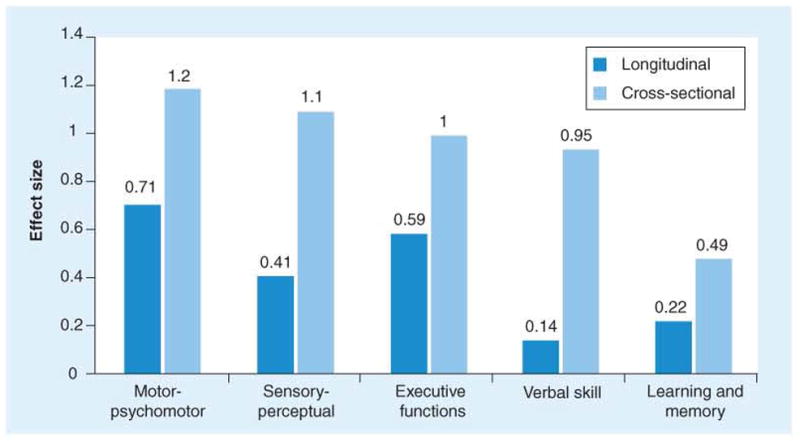
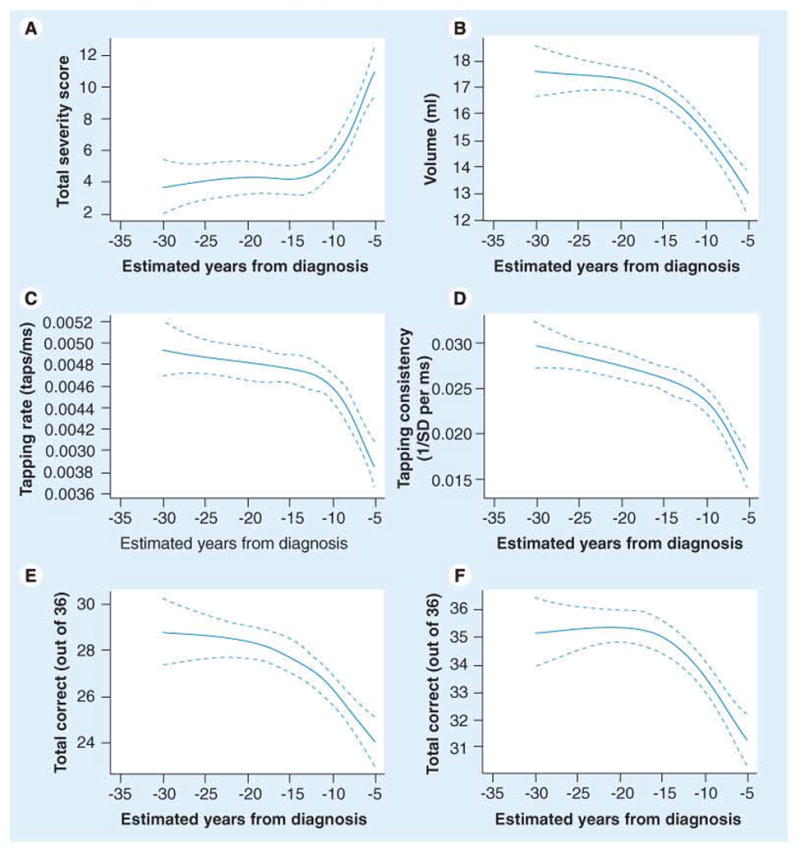
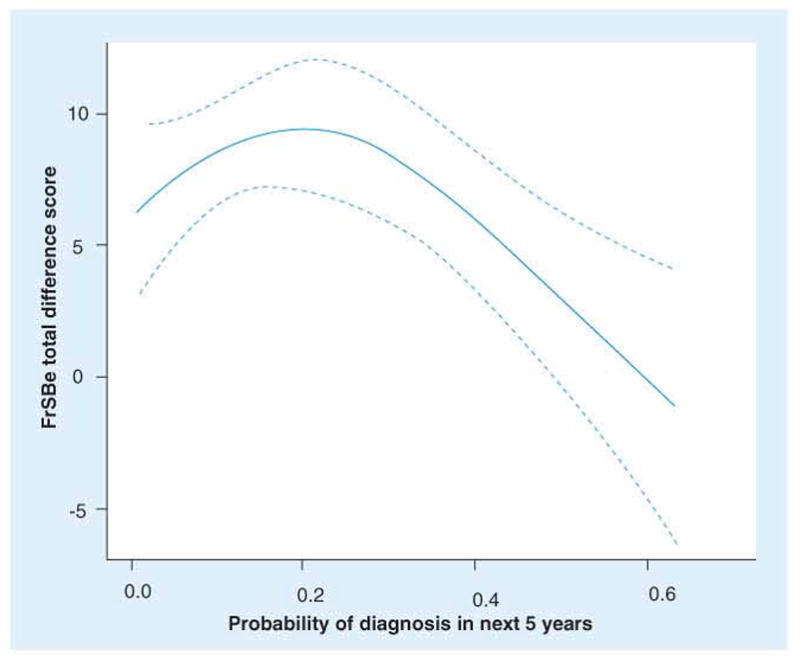
References
-
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–83. - PubMed
-
- Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington’s disease. Science. 1985;230:561–3. - PubMed
-
- Calabresi P, Centonze D, Pisani A, et al. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington’s disease. Ann Neurol. 1998;43:586–97. - PubMed
-
- Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntington’s disease. Neurosci Lett. 1991;133:257–61. - PubMed
-
- Paulsen JS, Mikos A. Huntington’s disease. In: Morgan JE, Ricker JH, editors. Textbook of clinical neuropsychology. Taylor & Francis; New York, NY: 2007. pp. 616–635.
Grants and funding
LinkOut - more resources
Full Text Sources