

MAN1B1 deficiency: an unexpected CDG-II
- PMID: 24348268
- PMCID: PMC3861123
- DOI: 10.1371/journal.pgen.1003989
MAN1B1 deficiency: an unexpected CDG-II
Abstract
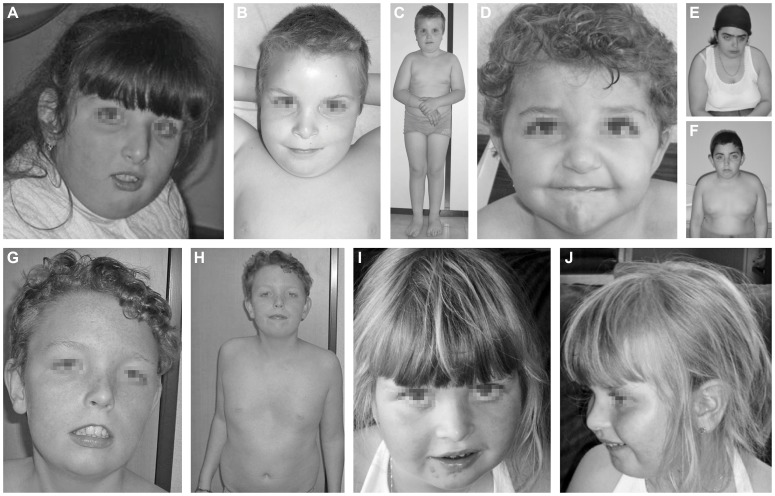
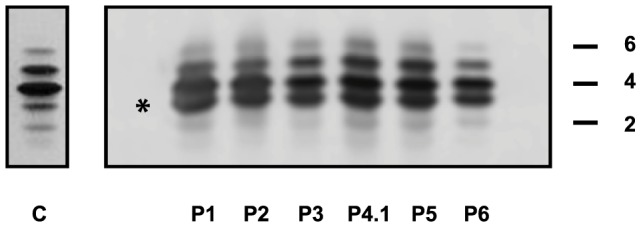
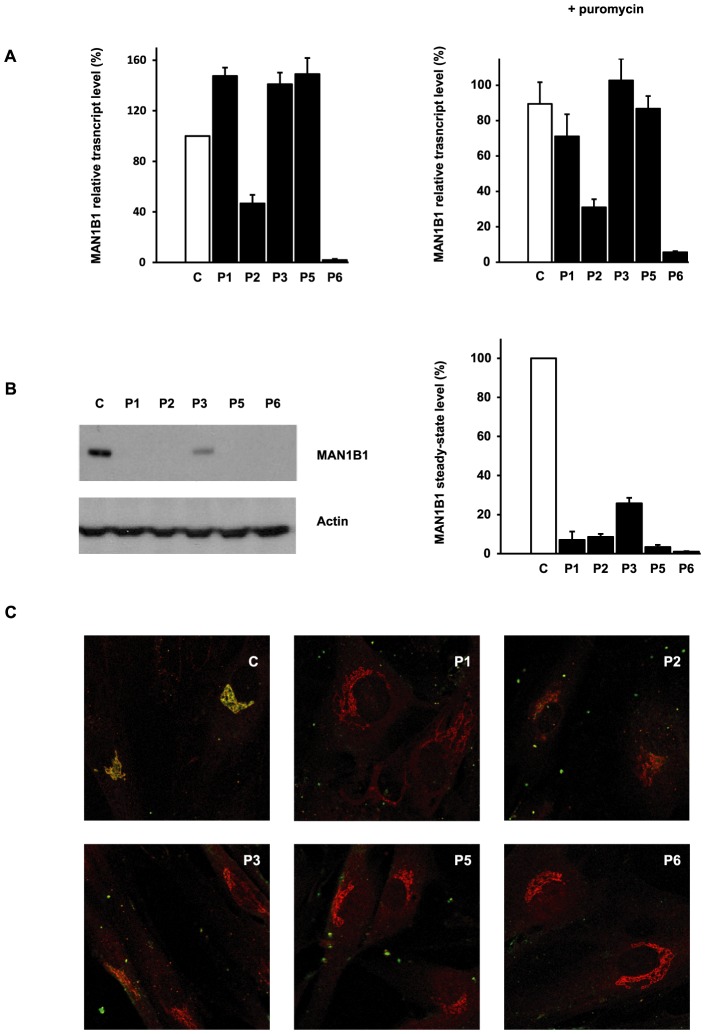
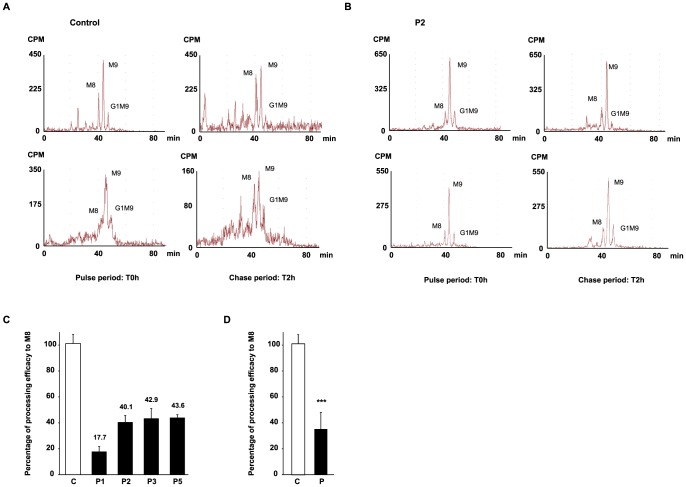
Congenital disorders of glycosylation (CDG) are a group of rare metabolic diseases, due to impaired protein and lipid glycosylation. In the present study, exome sequencing was used to identify MAN1B1 as the culprit gene in an unsolved CDG-II patient. Subsequently, 6 additional cases with MAN1B1-CDG were found. All individuals presented slight facial dysmorphism, psychomotor retardation and truncal obesity. Generally, MAN1B1 is believed to be an ER resident alpha-1,2-mannosidase acting as a key factor in glycoprotein quality control by targeting misfolded proteins for ER-associated degradation (ERAD). However, recent studies indicated a Golgi localization of the endogenous MAN1B1, suggesting a more complex role for MAN1B1 in quality control. We were able to confirm that MAN1B1 is indeed localized to the Golgi complex instead of the ER. Furthermore, we observed an altered Golgi morphology in all patients' cells, with marked dilatation and fragmentation. We hypothesize that part of the phenotype is associated to this Golgi disruption. In conclusion, we linked mutations in MAN1B1 to a Golgi glycosylation disorder. Additionally, our results support the recent findings on MAN1B1 localization. However, more work is needed to pinpoint the exact function of MAN1B1 in glycoprotein quality control, and to understand the pathophysiology of its deficiency.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



References
-
- Jaeken J, Matthijs G (2007) Congenital disorders of glycosylation: a rapidly expanding disease family. Annual review of genomics and human genetics 8: 261–278 Available: http://www.ncbi.nlm.nih.gov/pubmed/17506657. Accessed 2 February 2013. - PubMed
-
- Theodore M, Morava E (2011) Congenital disorders of glycosylation: sweet news. Current opinion in pediatrics 23: 581–587 Available: http://www.ncbi.nlm.nih.gov/pubmed/21970833. Accessed 7 February 2013. - PubMed
-
- Matthijs G, Rymen D, Millón MBB, Souche E, Race V (2013) Approaches to homozygosity mapping and exome sequencing for the identification of novel types of CDG. Glycoconjugate journal 30: 67–76 Available: http://www.ncbi.nlm.nih.gov/pubmed/22983704. Accessed 7 February 2013. - PubMed
-
- Stevens FJ, Argon Y (1999) Protein folding in the ER. Seminars in cell & developmental biology 10: 443–454 Available: http://www.ncbi.nlm.nih.gov/pubmed/10597627. Accessed 7 February 2013. - PubMed
-
- Jakob CA, Burda P, Roth J, Aebi M (1998) Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. The Journal of cell biology 142: 1223–1233 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2149342&tool=p.... Accessed 7 February 2013. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
