

Gene genealogies for genetic association mapping, with application to Crohn's disease
- PMID: 24348515
- PMCID: PMC3845011
- DOI: 10.3389/fgene.2013.00260
Gene genealogies for genetic association mapping, with application to Crohn's disease
Abstract
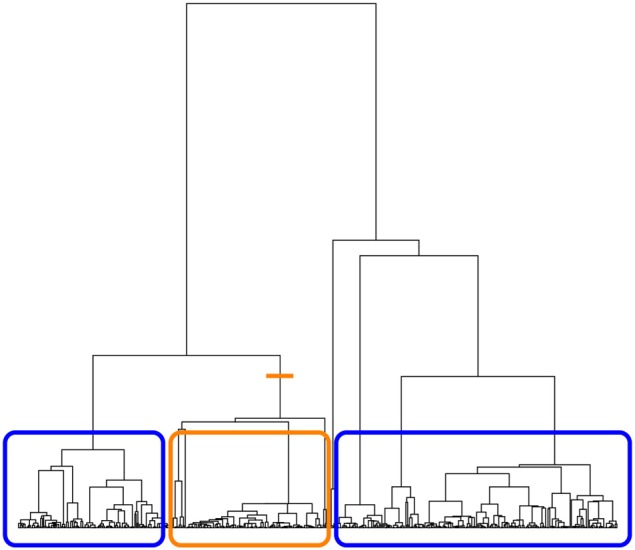
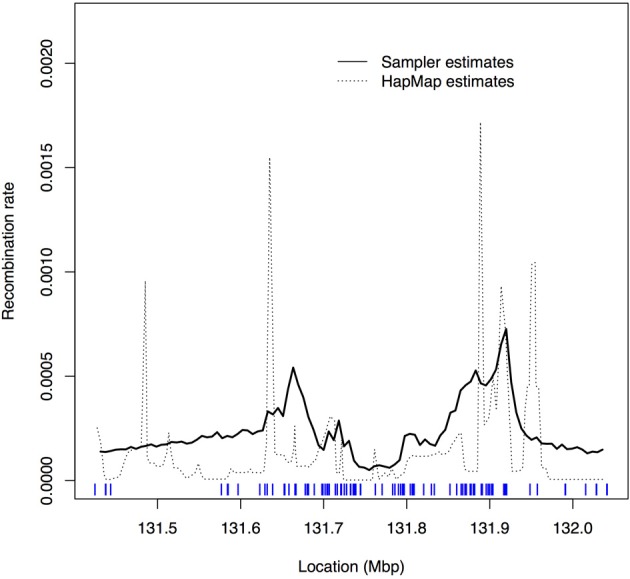
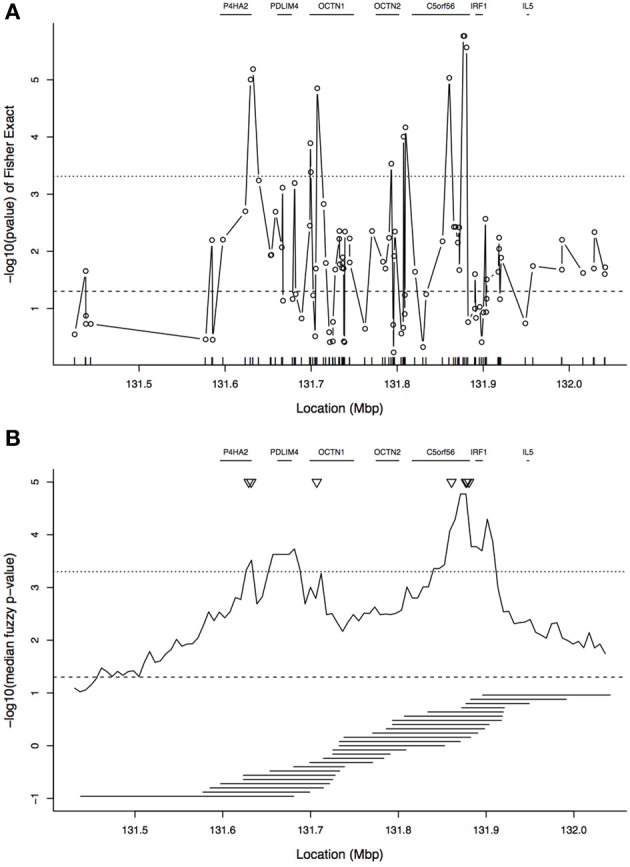
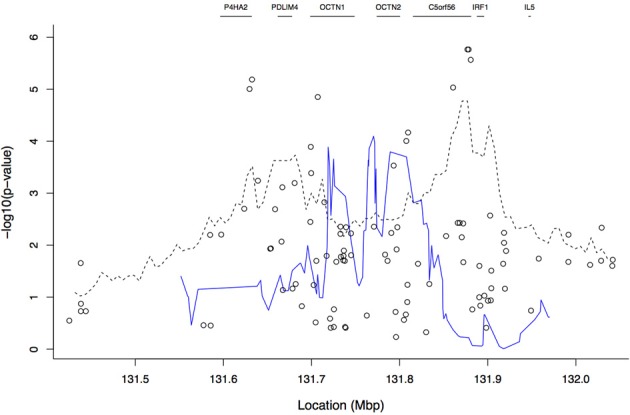
A gene genealogy describes relationships among haplotypes sampled from a population. Knowledge of the gene genealogy for a set of haplotypes is useful for estimation of population genetic parameters and it also has potential application in finding disease-predisposing genetic variants. As the true gene genealogy is unknown, Markov chain Monte Carlo (MCMC) approaches have been used to sample genealogies conditional on data at multiple genetic markers. We previously implemented an MCMC algorithm to sample from an approximation to the distribution of the gene genealogy conditional on haplotype data. Our approach samples ancestral trees, recombination and mutation rates at a genomic focal point. In this work, we describe how our sampler can be used to find disease-predisposing genetic variants in samples of cases and controls. We use a tree-based association statistic that quantifies the degree to which case haplotypes are more closely related to each other around the focal point than control haplotypes, without relying on a disease model. As the ancestral tree is a latent variable, so is the tree-based association statistic. We show how the sampler can be used to estimate the posterior distribution of the latent test statistic and corresponding latent p-values, which together comprise a fuzzy p-value. We illustrate the approach on a publicly-available dataset from a study of Crohn's disease that consists of genotypes at multiple SNP markers in a small genomic region. We estimate the posterior distribution of the tree-based association statistic and the recombination rate at multiple focal points in the region. Reassuringly, the posterior mean recombination rates estimated at the different focal points are consistent with previously published estimates. The tree-based association approach finds multiple sub-regions where the case haplotypes are more genetically related than the control haplotypes, and that there may be one or multiple disease-predisposing loci.
Keywords: Crohn's disease; Markov chain Monte Carlo; association study; coalescent model; fuzzy p-value; gene genealogy.
Figures
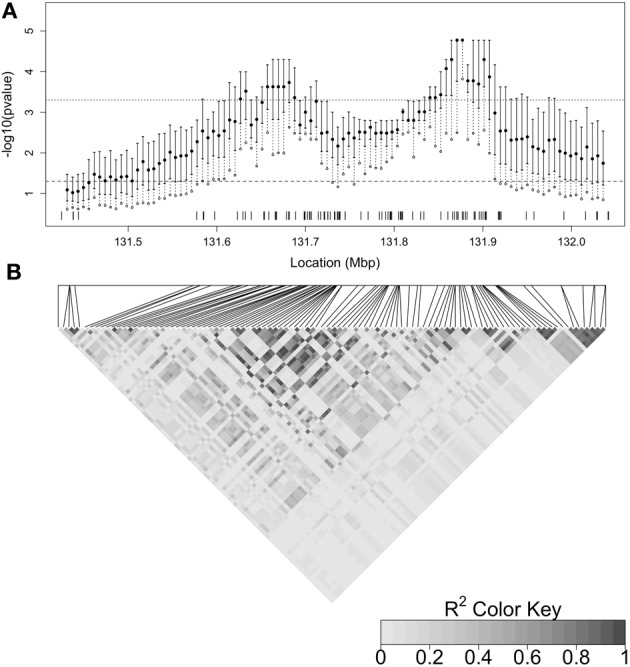
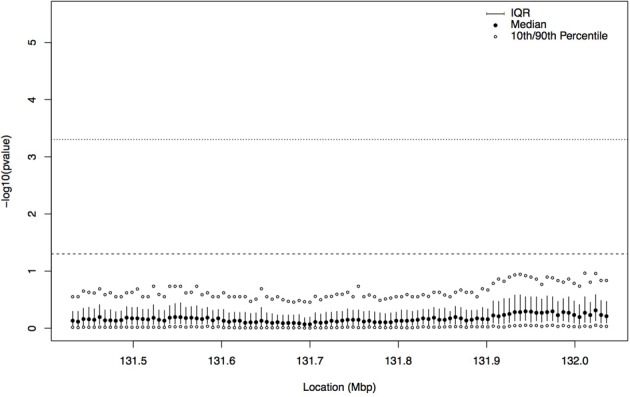
References
-
- Barrett M., Chandra S. B. (2011). A review of major Crohn's disease susceptibility genes and their role in disease pathogenesis. Genes Genom. 33, 317–325 10.1007/s13258-011-0076-3 - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials