

Large-scale production and structural and biophysical characterizations of the human hepatitis B virus polymerase
- PMID: 24352439
- PMCID: PMC3958071
- DOI: 10.1128/JVI.02575-13
Large-scale production and structural and biophysical characterizations of the human hepatitis B virus polymerase
Abstract
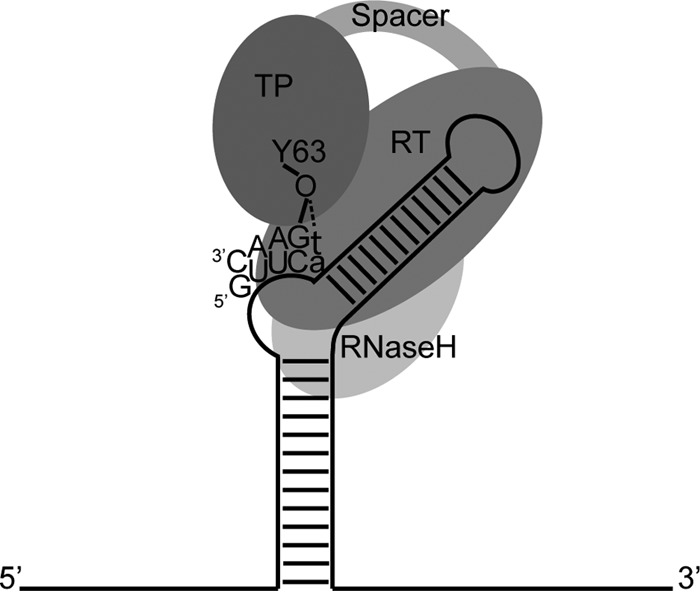
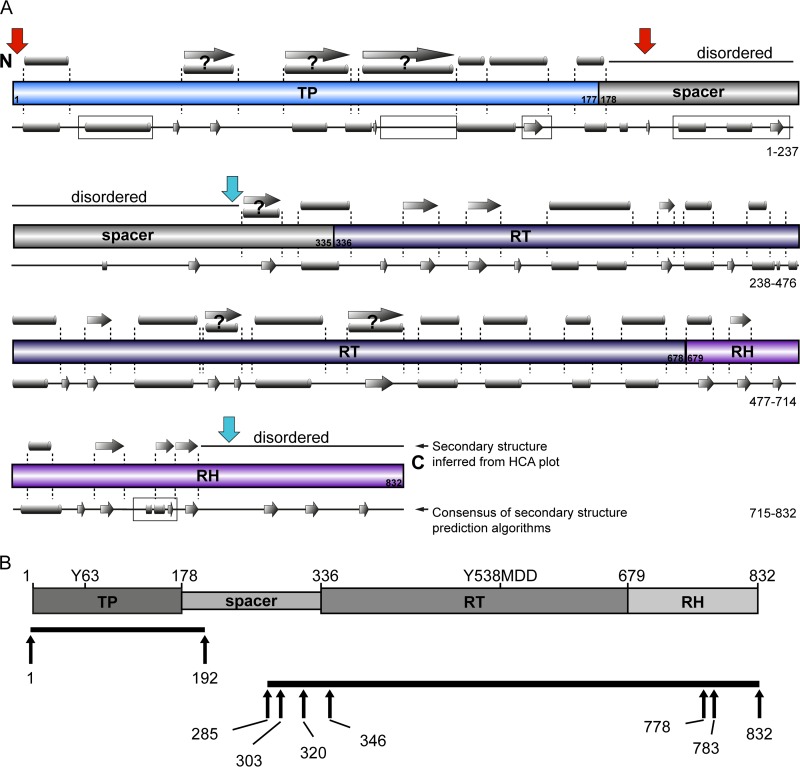
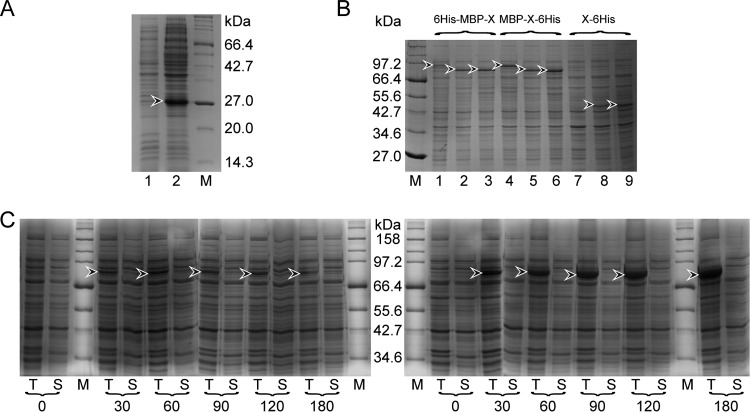
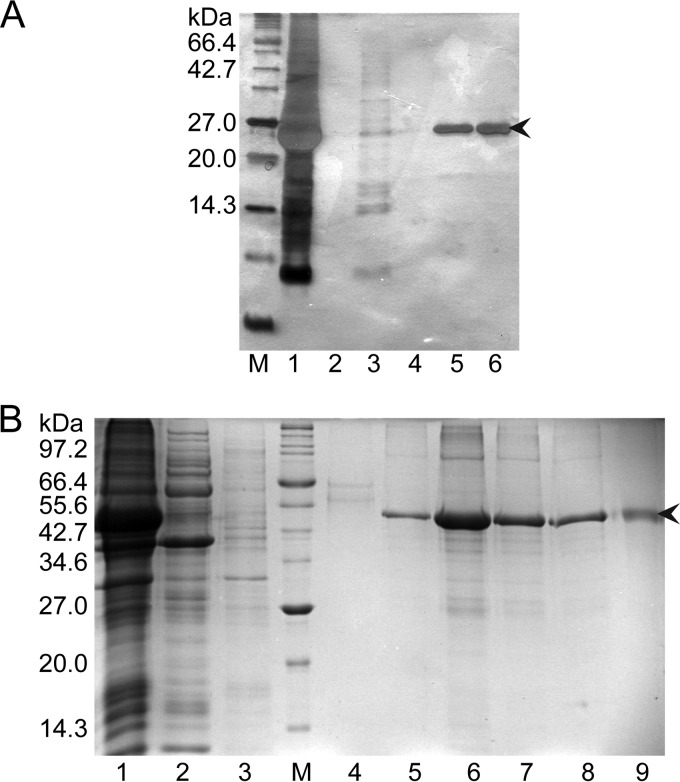
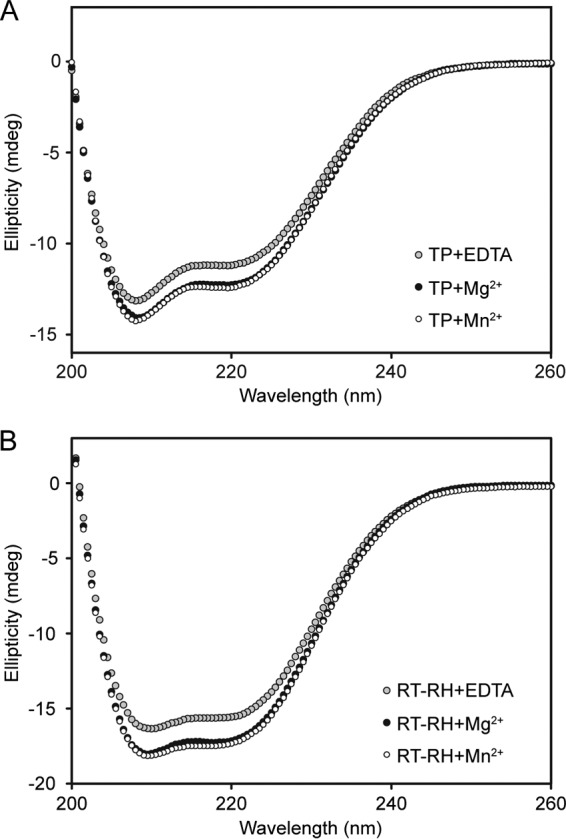
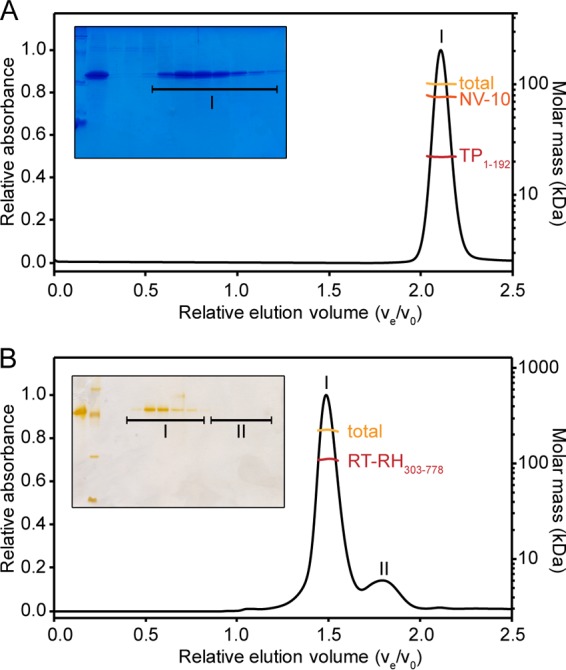
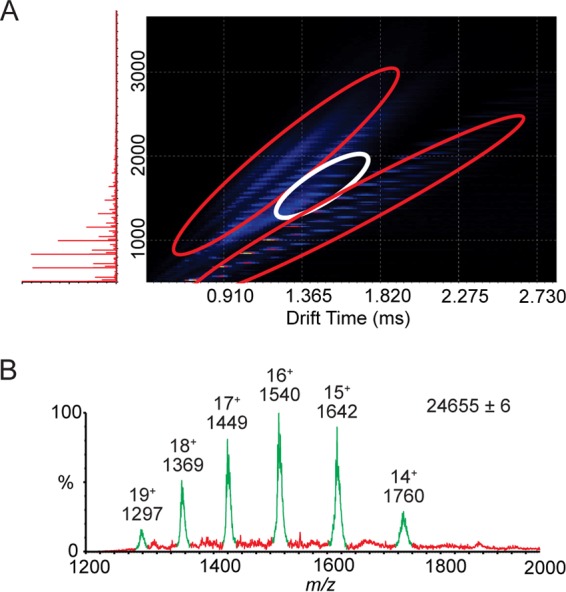
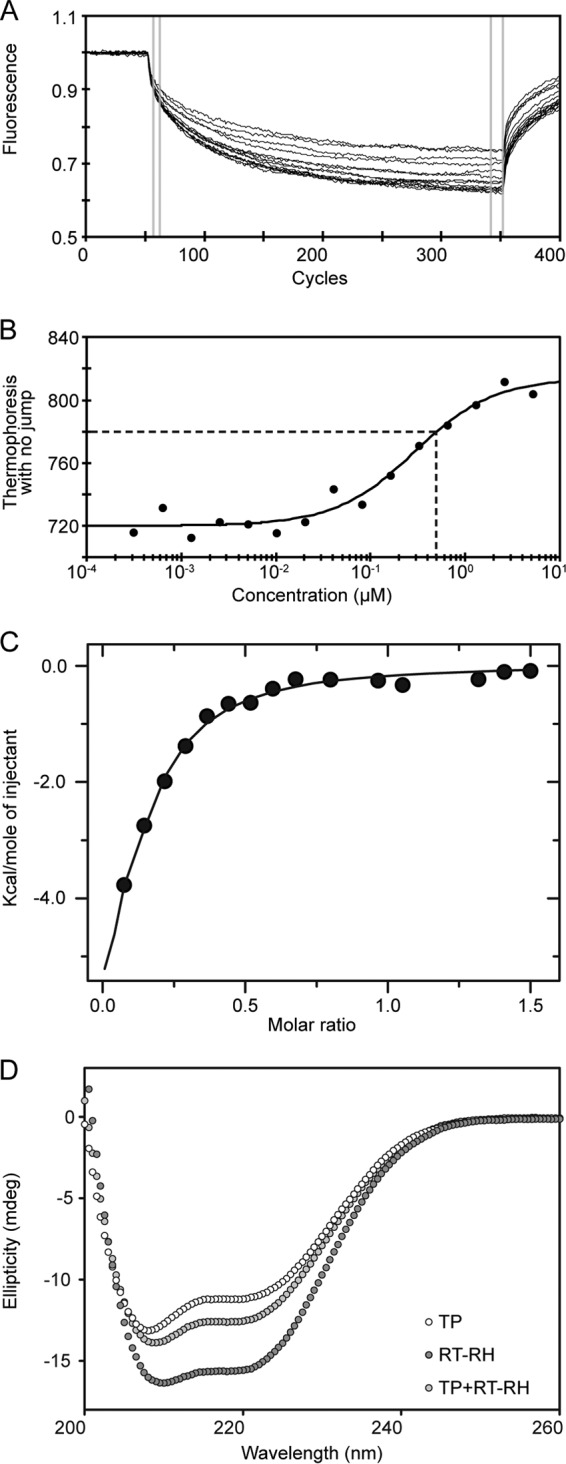
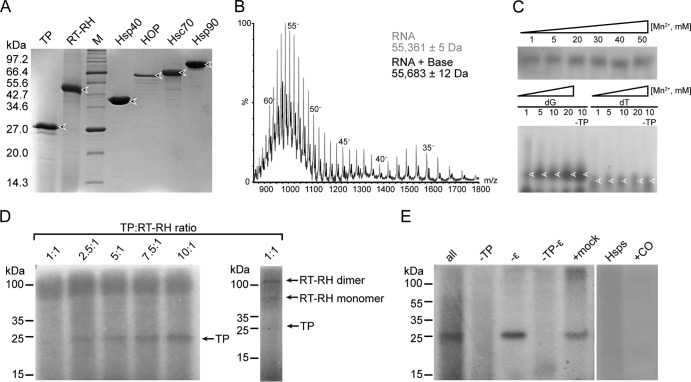
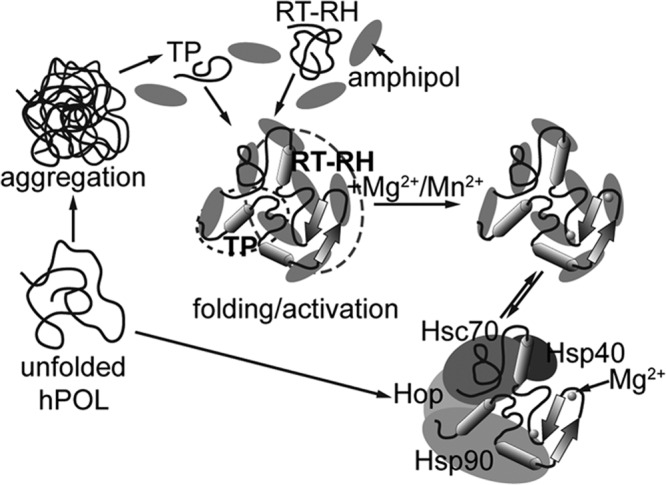
Hepatitis B virus (HBV) is a major human pathogen that causes serious liver disease and 600,000 deaths annually. Approved therapies for treating chronic HBV infections usually target the multifunctional viral polymerase (hPOL). Unfortunately, these therapies--broad-spectrum antivirals--are not general cures, have side effects, and cause viral resistance. While hPOL remains an attractive therapeutic target, it is notoriously difficult to express and purify in a soluble form at yields appropriate for structural studies. Thus, no empirical structural data exist for hPOL, and this impedes medicinal chemistry and rational lead discovery efforts targeting HBV. Here, we present an efficient strategy to overexpress recombinant hPOL domains in Escherichia coli, purifying them at high yield and solving their known aggregation tendencies. This allowed us to perform the first structural and biophysical characterizations of hPOL domains. Apo-hPOL domains adopt mainly α-helical structures with small amounts of β-sheet structures. Our recombinant material exhibited metal-dependent, reverse transcriptase activity in vitro, with metal binding modulating the hPOL structure. Calcomine orange 2RS, a small molecule that inhibits duck HBV POL activity, also inhibited the in vitro priming activity of recombinant hPOL. Our work paves the way for structural and biophysical characterizations of hPOL and should facilitate high-throughput lead discovery for HBV.
Importance: The viral polymerase from human hepatitis B virus (hPOL) is a well-validated therapeutic target. However, recombinant hPOL has a well-deserved reputation for being extremely difficult to express in a soluble, active form in yields appropriate to the structural studies that usually play an important role in drug discovery programs. This has hindered the development of much-needed new antivirals for HBV. However, we have solved this problem and report here procedures for expressing recombinant hPOL domains in Escherichia coli and also methods for purifying them in soluble forms that have activity in vitro. We also present the first structural and biophysical characterizations of hPOL. Our work paves the way for new insights into hPOL structure and function, which should assist the discovery of novel antivirals for HBV.
Figures
Similar articles
-
Expression and purification of a functional human hepatitis B virus polymerase.World J Gastroenterol. 2010 Dec 7;16(45):5752-8. doi: 10.3748/wjg.v16.i45.5752. World J Gastroenterol. 2010. PMID: 21128327 Free PMC article.
-
Mapping of Functional Subdomains in the Terminal Protein Domain of Hepatitis B Virus Polymerase.J Virol. 2017 Jan 18;91(3):e01785-16. doi: 10.1128/JVI.01785-16. Print 2017 Feb 1. J Virol. 2017. PMID: 27852858 Free PMC article.
-
Sequences in the terminal protein and reverse transcriptase domains of the hepatitis B virus polymerase contribute to RNA binding and encapsidation.J Viral Hepat. 2014 Dec;21(12):882-93. doi: 10.1111/jvh.12225. Epub 2014 Jan 9. J Viral Hepat. 2014. PMID: 24401091 Free PMC article.
-
Molecular, Evolutionary, and Structural Analysis of the Terminal Protein Domain of Hepatitis B Virus Polymerase, a Potential Drug Target.Viruses. 2020 May 22;12(5):570. doi: 10.3390/v12050570. Viruses. 2020. PMID: 32455999 Free PMC article. Review.
-
Hepatitis B viruses: reverse transcription a different way.Virus Res. 2008 Jun;134(1-2):235-49. doi: 10.1016/j.virusres.2007.12.024. Epub 2008 Mar 12. Virus Res. 2008. PMID: 18339439 Review.
Cited by
-
Preparation and functional evaluation of monoclonal antibodies targeting Hepatitis B Virus Polymerase.Virulence. 2021 Dec;12(1):188-194. doi: 10.1080/21505594.2020.1869391. Virulence. 2021. PMID: 33356842 Free PMC article.
-
Relaxing the restricted structural dynamics in the human hepatitis B virus RNA encapsidation signal enables replication initiation in vitro.PLoS Pathog. 2022 Mar 8;18(3):e1010362. doi: 10.1371/journal.ppat.1010362. eCollection 2022 Mar. PLoS Pathog. 2022. PMID: 35259189 Free PMC article.
-
Deviated binding of anti-HBV nucleoside analog E-CFCP-TP to the reverse transcriptase active site attenuates the effect of drug-resistant mutations.Sci Rep. 2024 Jul 8;14(1):15742. doi: 10.1038/s41598-024-66505-z. Sci Rep. 2024. PMID: 38977798 Free PMC article.
-
Structural features in common of HBV and HIV-1 resistance against chirally-distinct nucleoside analogues entecavir and lamivudine.Sci Rep. 2020 Feb 20;10(1):3021. doi: 10.1038/s41598-020-59775-w. Sci Rep. 2020. PMID: 32080249 Free PMC article.
-
The Impact of Mass Spectrometry-Based Proteomics on Fundamental Discoveries in Virology.Annu Rev Virol. 2014 Nov;1(1):581-604. doi: 10.1146/annurev-virology-031413-085527. Epub 2014 Jul 14. Annu Rev Virol. 2014. PMID: 26958735 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
