

doi: 10.1128/JVI.03333-13.
Epub 2013 Dec 18.
Epigenetic regulation of HIV-1 latency in astrocytes
Affiliations
- PMID: 24352441
- PMCID: PMC3958059
- DOI: 10.1128/JVI.03333-13
Item in Clipboard
Epigenetic regulation of HIV-1 latency in astrocytes
J Virol.
2014 Mar.
Abstract
HIV infiltrates the brain at early times postinfection and remains latent within astrocytes and macrophages. Because astrocytes are the most abundant cell type in the brain, we evaluated epigenetic regulation of HIV latency in astrocytes. We have shown that class I histone deacetylases (HDACs) and a lysine-specific histone methyltransferase, SU(VAR)3-9, play a significant role in silencing of HIV transcription in astrocytes. Our studies add to a growing body of evidence demonstrating that astrocytes are a reservoir for HIV.
Figures
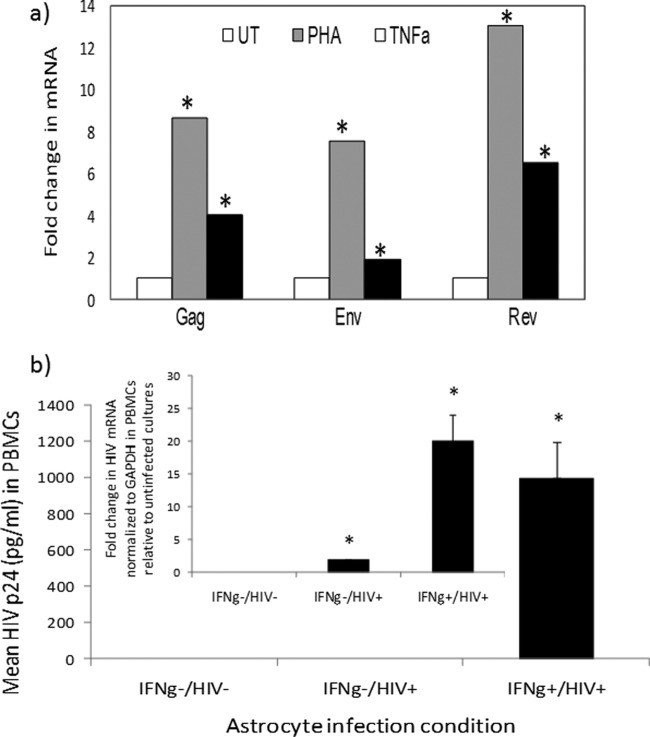
(a) Reactivation of HIV mRNAs in astrocytes. PDAs were infected with HIVBal (10 ng/ml/106 cells) and left untreated (UT) or treated with PHA (4 μg/ml) or TNF-α (TNFa) (100 U/ml), and HIV mRNAs were measured by qRT-PCR. Data are normalized to GAPDH mRNA and expressed as fold change relative to results for untreated cells. Asterisks denote P < 0.05 between results for treated and untreated cultures. (b) Inducible and low level of HIV replication in astrocytes is transmitted to lymphocytes. Astrocytes were primed with IFN-γ (75 U/ml) or left untreated and then infected with HIVBal (10 ng/ml/106 cells) and cultured with or without IFN-γ (IFNg). The initial virus inoculum was removed by mild pronase treatment and washing; the supernatant was harvested from astrocytes at day 7 postinfection and then exposed to anti-CD3/anti-CD28-costimulated PBMCs. HIV p24 from PBMC supernatant was measured by ELISA on day 6. HIV mRNA (Env) from PBMCs was quantified by real-time PCR on day 6, normalized to GAPDH, and presented as expression relative to that for uninfected cultures. HIV p24 from IFN-γ−/HIV− or HIV+ cultures was undetectable. *, P < 0.05 (Student's t test) between results for control and treated samples.

Estimation of frequency of HIV infection in astrocytes: a standard curve was generated to determine the percentage of infected cells based on use of a mixture of DNA from U1 (contains 2 proviral copies/cell) and U937 (U1 parental cell line). HIVBal (10 ng/ml/106 cells) was used to infect stimulated PBMCs, primary astrocytes (PDA), or an astrocytic cell line (U138). HIV DNA was amplified using Alu-PCR followed by real-time PCR. Linear regression list dotted line and standard curve are shown.

Transcript and protein expression profiles of HDACs in astrocytes. (a) RNA extracts (1 μg) from PDAs were subjected to real-time PCR to detect and quantify all of the 11 HDACs and CCDN1. GAPDH was used as an internal control, and data are presented as fold change relative to results for CCDN1. (b and c) Protein extracts (20 μg each) from PDAs (b) or U87MG (c) were used to perform Western blot analysis using antibodies specific to HDAC1, -2, -3, and -4 from an HDAC antibody sampling kit (Cell Signaling) or GAPDH. *, P < 0.05 (Student's t test) between results for CCDN1 and HDAC2.

HDAC class 1 inhibitors significantly induce HIV LTR activity in astrocytes: U87MG cells with stably integrated HIV LTR-Luc were treated with various concentrations of HDAC inhibitors having selective specificity as indicated in Table 1. The agents were SAHA (a), apicidin (b),TSA (c), VAHA (d), MC1568 (e), salermide (f), splitomicin (g), or 1-naphthohydroxamic acid (h). Approximately 24 h posttreatment, cells were lysed and measured for luciferase activity. The data are normalized to total protein content and presented as fold change with respect to results for DMSO-treated cells ± SD. *, P < 0.05 (Student's t test) between results for DMSO-treated and drug-treated samples.

Induction of stable LTR activity is specific to trimethylation of histone H3 at Lys9 (H3K9me3). LTR-Luc-stably integrated U87MG cells were treated with AZA, a DNMT inhibitor (a), garcinol, a potent HAT inhibitor (b), UNC0638, GLP (H3K9 monomethyltransferase) and G9a (H3K9 dimethyltransferase) inhibitor (c), BIX0124, a specific G9a inhibitor (d), or chaetocin, a specific inhibitor of SU(VAR)3–9, an H3K9 trimethyltransferase (e), at the concentrations indicated. Approximately 24 h posttreatment, the cells were lysed and assayed for luciferase activity, normalized to total protein content. Data are presented as fold change with respect to results for DMSO-treated cells ± SD. *, P < 0.05 (Student's t test) between DMSO-treated and drug-treated samples.

SAHA, apicidin, and chaetocin strongly induce HIV transcripts and infectious virus from PDAs and U138MG astrocytes. PDAs (a) or U138MG cells (b) were infected with HIVBal and treated with SAHA (10 μM), apicidin (1 μg/ml), chaetocin (200 nm), or vehicle control. RNA was isolated 24 h posttreatment, 1 μg reverse transcribed to cDNA, and subjected to real-time PCR to quantify HIV Gag, Env, and Rev (c) transcripts. Data are normalized to results for GAPDH and are represented as fold change with respect to results for control (DMSO)-treated cells ± SD. (d) Supernatants from SAHA-, chaetocin-, or DMSO-treated HIVBal-infected PDAs were added to anti-CD3/anti-CD28-activated PBMCs. At day 6, HIV transcript (Env) from PBMCs was quantified by real-time PCR. Data are normalized to results for GAPDH and are represented as fold change with respect to results for control-treated samples. *, P < 0.05 (Student's t test) between results for control (DMSO)-treated and inhibitor-treated samples.
References
-
- Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, O'Shea A, Callender M, Spivak A, Brennan T, Kearney MF, Proschan MA, Mican JM, Rehm CA, Coffin JM, Mellors JW, Siliciano RF, Maldarelli F. 2009. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. U. S. A. 106:9403–9408. 10.1073/pnas.0903107106 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
