

Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation
- PMID: 24352472
- PMCID: PMC3958050
- DOI: 10.1128/JVI.02727-13
Hepatitis C virus stimulates low-density lipoprotein receptor expression to facilitate viral propagation
Abstract
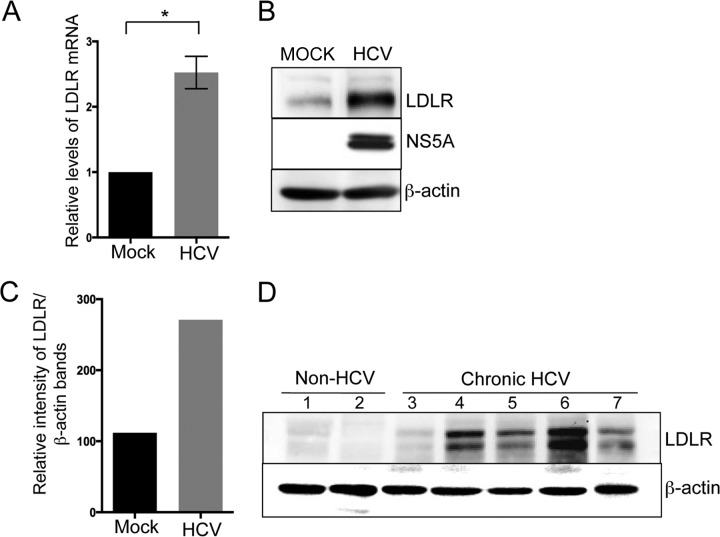
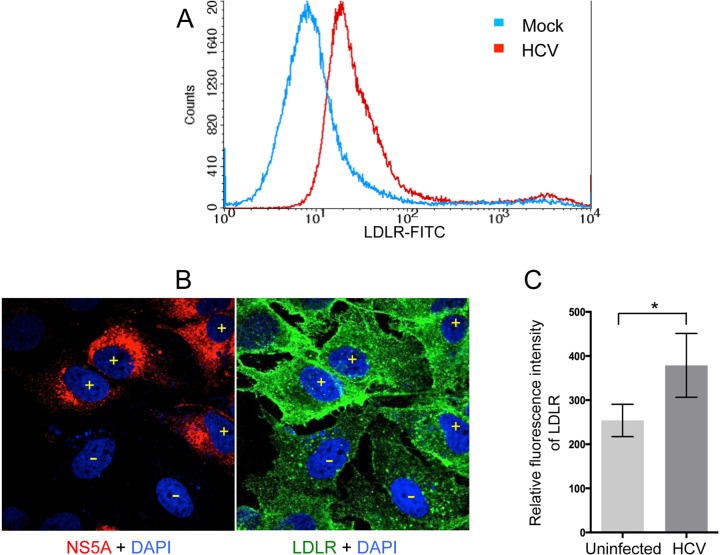
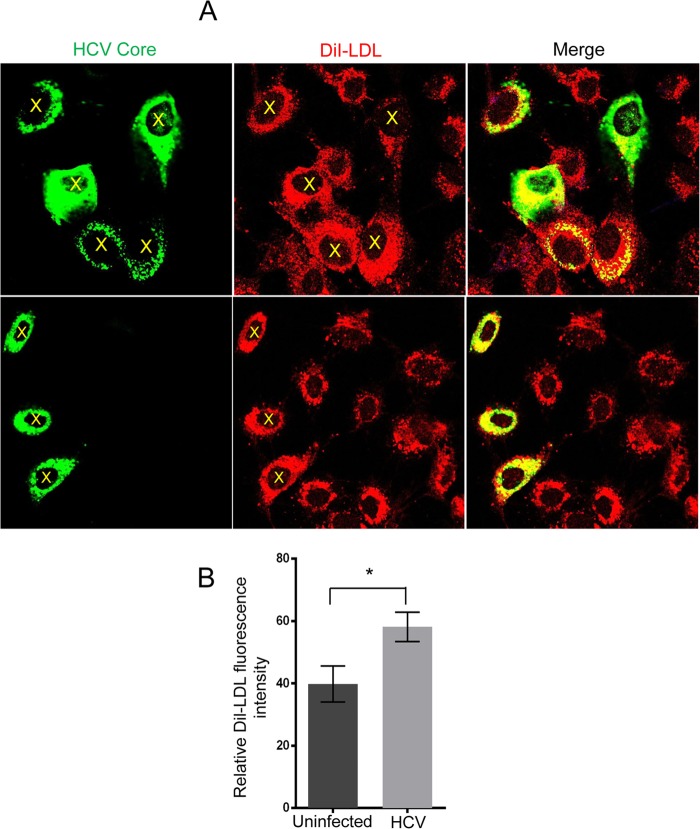
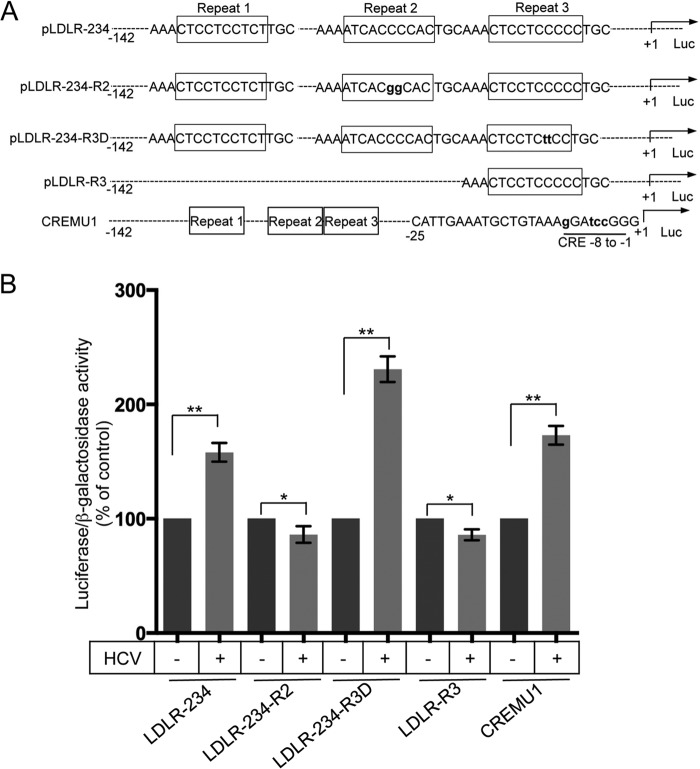
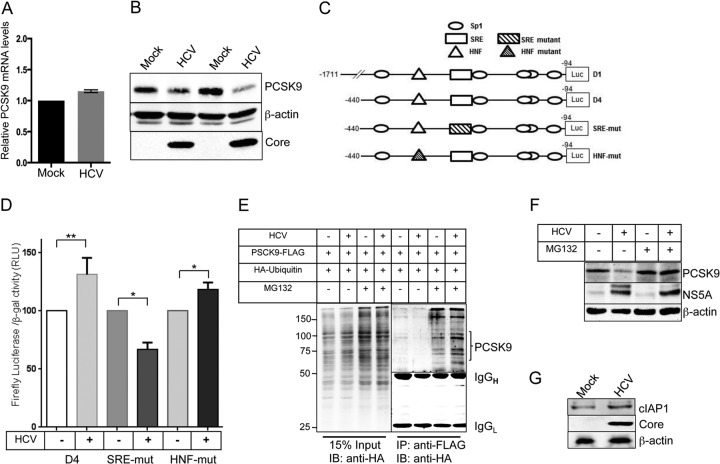
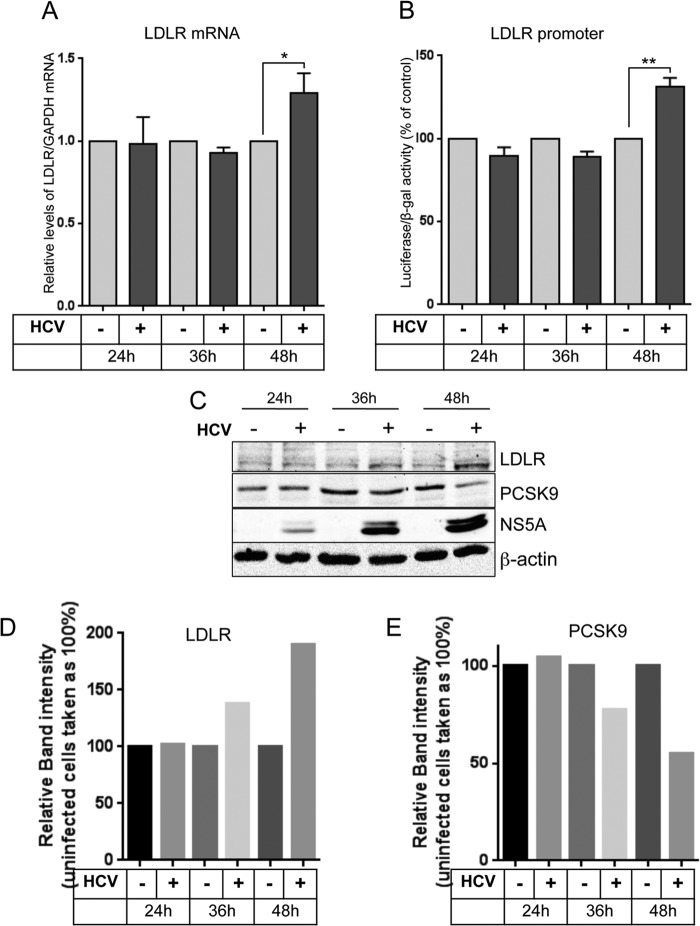
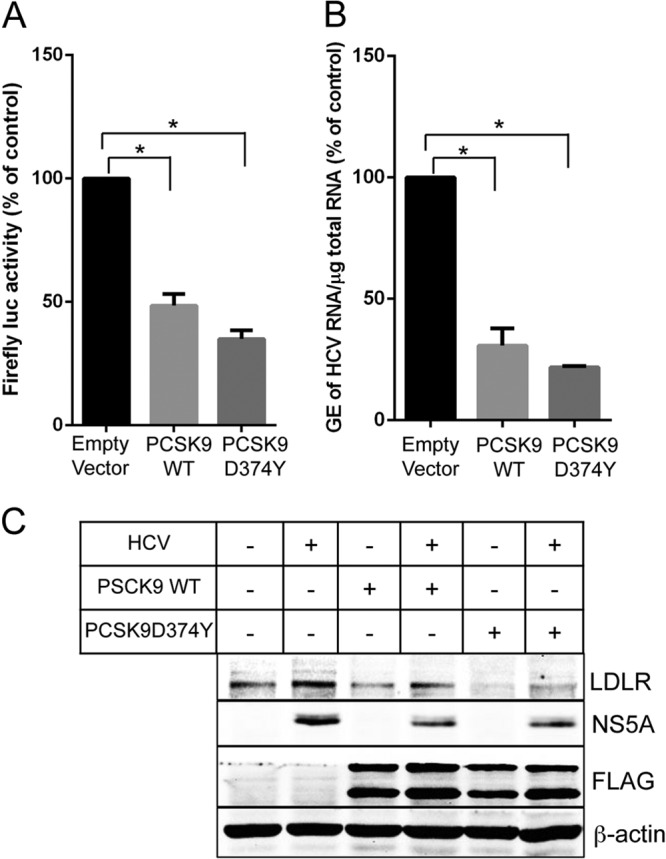
Lipids play a crucial role in multiple aspects of hepatitis C virus (HCV) life cycle. HCV modulates host lipid metabolism to enrich the intracellular milieu with lipids to facilitate its proliferation. However, very little is known about the influence of HCV on lipid uptake from bloodstream. Low-density lipoprotein receptor (LDLR) is involved in uptake of cholesterol rich low-density lipoprotein (LDL) particles from the bloodstream. The association of HCV particles with lipoproteins implicates their role in HCV entry; however, the precise role of LDLR in HCV entry still remains controversial. Here, we investigate the effect of HCV infection on LDLR expression and the underlying mechanism(s) involved. We demonstrate that HCV stimulates LDLR expression in both HCV-infected Huh7 cells and in liver tissue from chronic hepatitis C patients. Fluorescence activated cell sorting and immunofluorescence analysis revealed enhanced cell surface and total expression of LDLR in HCV-infected cells. Increased LDLR expression resulted in the enhanced uptake of lipoprotein particles by HCV-infected cells. Analysis of LDLR gene promoter identified a pivotal role of sterol-regulatory element binding proteins (SREBPs), in the HCV-mediated stimulation of LDLR transcription. In addition, HCV negatively modulated the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9), a protein that facilitates LDLR degradation. Ectopic expression of wild-type PCSK9 or gain-of-function PCSK9 mutant negatively affected HCV replication. Overall, our results demonstrate that HCV regulates LDLR expression at transcriptional and posttranslational level via SREBPs and PCSK9 to promote lipid uptake and facilitate viral proliferation.
Importance: HCV modulates host lipid metabolism to promote enrichment of lipids in intracellular environment, which are essential in multiple aspects of HCV life cycle. However, very little is known about the influence of HCV on lipid uptake from the bloodstream. LDLR is involved in uptake of cholesterol rich lipid particles from bloodstream. In this study, we investigated the effect of HCV on LDLR expression and the underlying mechanism triggered by the virus to modulate LDLR expression. Our observations suggest that HCV upregulates LDLR expression at both the protein and the transcript levels and that this upregulation likely contributes toward the uptake of serum lipids by infected hepatocytes. Abrogation of HCV-mediated upregulation of LDLR inhibits serum lipid uptake and thereby perturbs HCV replication. Overall, our findings highlight the importance of serum lipid uptake by infected hepatocytes in HCV life cycle.
Figures
Similar articles
-
Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis.J Am Coll Cardiol. 2013 Oct 15;62(16):1401-8. doi: 10.1016/j.jacc.2013.07.056. Epub 2013 Aug 21. J Am Coll Cardiol. 2013. PMID: 23973703 Review.
-
PCSK9, apolipoprotein E and lipoviral particles in chronic hepatitis C genotype 3: evidence for genotype-specific regulation of lipoprotein metabolism.J Hepatol. 2015 Apr;62(4):763-70. doi: 10.1016/j.jhep.2014.11.016. Epub 2014 Nov 21. J Hepatol. 2015. PMID: 25463543
-
Peroxisome Proliferator-activated receptor γ activation by ligands and dephosphorylation induces proprotein convertase subtilisin kexin type 9 and low density lipoprotein receptor expression.J Biol Chem. 2012 Jul 6;287(28):23667-77. doi: 10.1074/jbc.M112.350181. Epub 2012 May 16. J Biol Chem. 2012. PMID: 22593575 Free PMC article.
-
β-Estradiol results in a proprotein convertase subtilisin/kexin type 9-dependent increase in low-density lipoprotein receptor levels in human hepatic HuH7 cells.FEBS J. 2015 Jul;282(14):2682-96. doi: 10.1111/febs.13309. Epub 2015 May 18. FEBS J. 2015. PMID: 25913303 Free PMC article.
-
PCSK9 and LDLR degradation: regulatory mechanisms in circulation and in cells.Curr Opin Lipidol. 2014 Oct;25(5):387-93. doi: 10.1097/MOL.0000000000000114. Curr Opin Lipidol. 2014. PMID: 25110901 Free PMC article. Review.
Cited by
-
New Mechanism of Hepatic Fibrogenesis: Hepatitis C Virus Infection Induces Transforming Growth Factor β1 Production through Glucose-Regulated Protein 94.J Virol. 2015 Dec 30;90(6):3044-55. doi: 10.1128/JVI.02976-15. J Virol. 2015. PMID: 26719248 Free PMC article.
-
Statins and PCSK9 inhibitors: What is their role in coronavirus disease 2019?Med Hypotheses. 2021 Jan;146:110452. doi: 10.1016/j.mehy.2020.110452. Epub 2020 Dec 9. Med Hypotheses. 2021. PMID: 33333472 Free PMC article. Review.
-
Transcriptome and miRNome Analysis Provide New Insight Into Host Lipid Accumulation, Innate Immunity, and Viral Persistence in Hepatitis C Virus Infection in vitro.Front Microbiol. 2020 Sep 30;11:535673. doi: 10.3389/fmicb.2020.535673. eCollection 2020. Front Microbiol. 2020. PMID: 33101221 Free PMC article.
-
Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and Beyond.Int J Mol Sci. 2022 Jan 19;23(3):1070. doi: 10.3390/ijms23031070. Int J Mol Sci. 2022. PMID: 35162992 Free PMC article. Review.
-
Insight into the Evolving Role of PCSK9.Metabolites. 2022 Mar 17;12(3):256. doi: 10.3390/metabo12030256. Metabolites. 2022. PMID: 35323699 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
